Cannabis Metabolomic Data Processing: Challenges to be Addressed
The interest in cannabis products is growing exponentially, because they are expected to provide a number of beneficial and therapeutic effects. Cannabis plants contain a number of unique biologically active secondary metabolites, with their pattern largely dependent not only on a respective variety or chemotype but also on several other factors. This article presents the challenges that arise during cannabis metabolomics analysis using ultrahigh-performance reversed‑phase liquid chromatography coupled with high-resolution tandem mass spectrometry (UHPLC–reversed‑phase–HRMS/MS).
For this purpose, ethanolic extracts of two cannabis varieties were measured twice within one month and the obtained datasets were chemometrically analyzed with respect to the analysis date. Rather surprisingly, the effect of time variability was observed at the level of expression of relative abundance of features as well as in multivariate models, where not only variety clustering but also time-dependent clustering occurred. The data handling workflow, within time variability can be revealed and removed by advanced normalization method, is described.
Cannabis sativa L. (Cannabaceae) is an annual plant that has been used for its therapeutic and other effects for a thousand years. Up to now, more than 750 secondary metabolites have been identified in cannabis plants, with over 120 of them representing unique C21 terpenophenolic constituents named phytocannabinoids (1–3). Tetrahydrocannabivarin, cannabinol, cannabidiol (CBD), cannabidivarin, cannabigerol (CBG), delta-9-tertrahydrocannabinol (Δ9-THC), and cannabichromene with its acidic forms (the respective carboxylic acids) are currently of major concern when assessing cannabis quality. The other bioactive metabolites that occur in a complex mixture belong to flavonoids, alkaloids, stilbenoids, and phenols (4). It should be noted that the cannabis metabolome is highly variable, depending not only on the respective variety or chemotype (5) but also on growing conditions (6). Moreover, cannabis post-harvest processing, including drying, extraction, and other practices may have a significant impact on product composition and thus overall biological activity. Although such variability offers diversity in their uses, unexpected treatment effects should be taken into consideration as well (7). In any case, effective tools for fit‑to‑purpose laboratory control of cannabis-based products should be available.
Currently, gas chromatography (GC) is the method of choice for the characterization of volatile terpenes in cannabis and products thereof (8). For a long time, it also used to be the primary analytical method for major phytocannabinoids quantification (9,10). However, due to the thermolability of acidic forms (their decarboxylation to neutral forms takes place at elevated temperatures), time‑consuming derivatization should be used for accurate analysis. As these limitations are not encountered in liquid chromatography (LC), this technique has become the gold standard in this field (11). Recently developed methods using ultrahigh‑performance liquid chromatography (UHPLC) coupled to diode-array detection (DAD) or mass spectrometry (MS) make reliable quantification of up to 18 major phytocannabinoids (using analytical standards available for the compounds mentioned above) in various matrices possible (12–14). However, considering the complexity of the cannabis metabolome and the limited stability of some components, more comprehensive information is needed to characterize cannabis samples. Target analysis of major phytocannabinoids was shown to be insufficient for chemotypes classification (based on dominating components Δ9-THC, CBD, or CBG), and so untargeted metabolomics was investigated by several research teams (15). “Phytocannabinomics” appears to be a promising approach, enabling the discovery of cannabis chemovars and varieties with unique phytocannabinoid profiles (16). Although UHPLC coupled to high-resolution mass spectrometry (HRMS) or tandem mass spectrometry (MS/MS) accounts for a significant proportion of applications (17,18,19), nuclear magnetic resonance (NMR) (20,21) or GC coupled to a flame ionization detector (FID) (22) can serve as complementary techniques too.
Performed cannabis analytical characterization can be divided into three main approaches: target analyses, metabolomics (targeted) profiling, and (nontarget) fingerprinting (23). When developing cannabis analyses, one has to deal not only with matrix effects but also simultaneous determination of abundant and minor cannabinoids (24). The advantage of target analyses is the quantification based on the calibration curve of the analytical standards (including internal standards), which is performed after each completed sequence, and the absolute concentrations supplemented by uncertainties are independent of any subsequent analyses, and thus can be easily compared. The opposite situation occurs for screening and metabolomics‑type analyses, where the unavailability of analytical standards makes the transfer of peak intensities to any reference value impossible, and so these results are usually expressed as relative abundances. This relativization is called total ion current (TIC) normalization and is the most common normalization technique. The process of converting the absolute intensities to relative ones by forcing all the sample to have equal total intensity aims to eliminate sources of systematic variation between sample profiles. TIC normalization works well, until the ion intensities exceed the linear range of detector and inconsistent nonlinear changes are observed across peaks (25). However, during LC–MS measurement, specific deviations may occur, which
differ between features. Thus, an algorithm called Quality Control‑Robust LOESS Signal Correction (QC‑RLSC), developed by Dunn et al., was introduced. During QC‑RLSC, a correction curve is interpolated for each feature through the whole analytical run to which the feature is normalized (26). Quality control (QC) samples are usually created by pooling aliquots of all samples in sequence and injecting them during the whole analytical run at the same intervals (every 10 injections) (27). As LC–MS measurement is batch-wise, such prepared QCs can be used as a reference level between batches using the same normalization correction procedure, QC-RLSC (28).
Metabolomics analysis generates big data and their processing should be carefully performed to obtain unbiased interpretation.
This study presents a solution to a problem encountered when handling the data obtained by UHPLC–HRMS/MS analysis of cannabis extracts of two varieties in different time slots. The importance of choosing the relevant chemometric tools for the correct results interpretation is emphasized.
Materials and Methods
Chemicals: All of the LC–MS-grade chemicals (ethanol [EtOH], acetonitrile, 2-propanol [iPrOH]) and mobile phase modifiers (ammonium acetate, acetic acid) were purchased from Merck. Deionized water (H2O) was obtained from a Milli-Q Integral system (Millipore).
Sample Preparation: For the experiment, 30 samples of dried and homogenized cannabis plants (15 samples of Santhica variety and 15 samples of Bialobrzeskie variety, harvested during July–October 2016 in Olomouc and Sumperk locality) were used. A representative sample (0.5 g) was weighed into a centrifuge tube (50 mL), mixed with 20 mL of EtOH, and shaken for 30 min at 240 rpm. The extracts were centrifugated (5 min, 10,000 rpm) and filtered into a 50 mL flask. The solid residue was extracted again using the same extraction procedure and the final pooled extract was made up to 50 mL. Aliquots were transferred into vials for UHPLC–HRMS/MS analysis. All samples were measured in duplicates (in randomized order). QC samples were prepared by pooling 100 µL of each sample and were run together with all the samples in the beginning of the sequence and then inserted after each series of 10 samples in order to check instrument conditions and control the sequence during measurement. Blank samples contained EtOH only.
Instrumental Analysis: For the metabolomics analysis, an UHPLC system (Infinity 1290, Agilent) coupled with a high-resolution tandem mass spectrometer (6560 Ion Mobility QTOF LC–MS, Agilent) was employed. Chromatographic separation was performed using a 2.1 × 150 mm, 1.7-μm Acquity BEH C18 column (Waters). The mobile phase consisted of (A) 5 mM ammonium acetate in a mixture of 95:5 H2O–acetonitrile (v/v) with 0.1% acetic acid and (B) 5 mM ammonium acetate in a mixture of 75:20:5 iPrOH–acetonitrile–H2O (v/v/v) with 0.1% acetic acid and the following elution gradient was used: 0.0 min (100% A, 0.3 mL/min), 0.5 min (100% A, 0.3 mL/min), 4.0 min (35% A, 0.3 mL/min), 4.01 min (35% A, 0.2 mL/min), 8.0 min (22.5% A, 0.2 mL/min), 13.0 min (0% A, 0.2 mL/min), 13.01 min (0% A, 0.35 mL/min), 18.0 min (0% A, 0.350 mL/min), 18.01 min (100% A, 0.3 mL/min), 20.0 min (100% A, 0.3 min/mL). The column temperature was maintained at 60 °C, the injection volume was set to 1 µL. The autosampler was kept at 10°C. The mass spectrometer was operated in the positive ionization mode (ESI+)/negative ionization mode (ESI-) using auto MS/MS acquisition mode in the mass range 100–1000 mass-to-charge ratio (m/z) under the following conditions: gas temperature, 280/300 °C; drying gas, 12 L/min; nebulizer pressure, 40/25 psig; sheath gas temperature, 350/370 °C; sheath gas flow, 12 L/min; nozzle voltage, 400 V; fragmentor voltage, 380 V; octopole radiofrequency voltage, 750 V; capillary voltage, 3500 V.
Design of Experiment: Two Cannabis sativa L. varieties—Santhica and Bialobrzeskie—were selected for the study. Fifteen samples of each variety were repeatedly extracted with EtOH (according to the procedure described above) and two aliquots of each sample were transferred into vials. A QC sample was prepared as a pool of all samples (100 μL of each sample) and two aliquots were made. The first batch (first aliquots) was measured immediately, the second batch was stored for 2 weeks at -80 °C.
Data Processing and Statistical Evaluation: The acquired primary data were processed with MZmine 2, where QC samples were used for feature extraction and acquired mass lists were used for targeted feature extraction in all other samples. The threshold for minimum peak height was set to 10,000. Data matrices for each ionization mode including features characterized by retention time, m/z, and intensity in each sample were created. Statistical analysis was performed in Metaboanalyst (metaboanalyst.ca) and using custom-built R scripts. Prior to any statistical analysis, sum normalization and logarithmic transformation were performed. In the case of multivariate chemometric models, Pareto scaling was applied for signal processing. QC-RLSC normalization was performed using in‑house R script. For time variability, paired t-tests were used on significance level α = 0.01 using Benjamini Hochberg multiple testing correction (false discovery rate [FDR]). After Pareto scaling and mean centring, the data were overviewed using principal component analysis (PCA).
Results and Discussion
As mentioned earlier, metabolomic fingerprinting represents a challenging tool for a comprehensive characterization of cannabis samples and their authentication. The working plan was to analyze samples (ethanolic extracts obtained from two sets of cannabis varieties, Santhica and Bialobrzeskie) two times to control the variability of measurements. The data obtained by the first and second measurement, which was realized three weeks later, were processed together, and after alignment the final data matrix contained 834 features: 290 and 544 features were detected in ESI+ and ESI-, respectively.
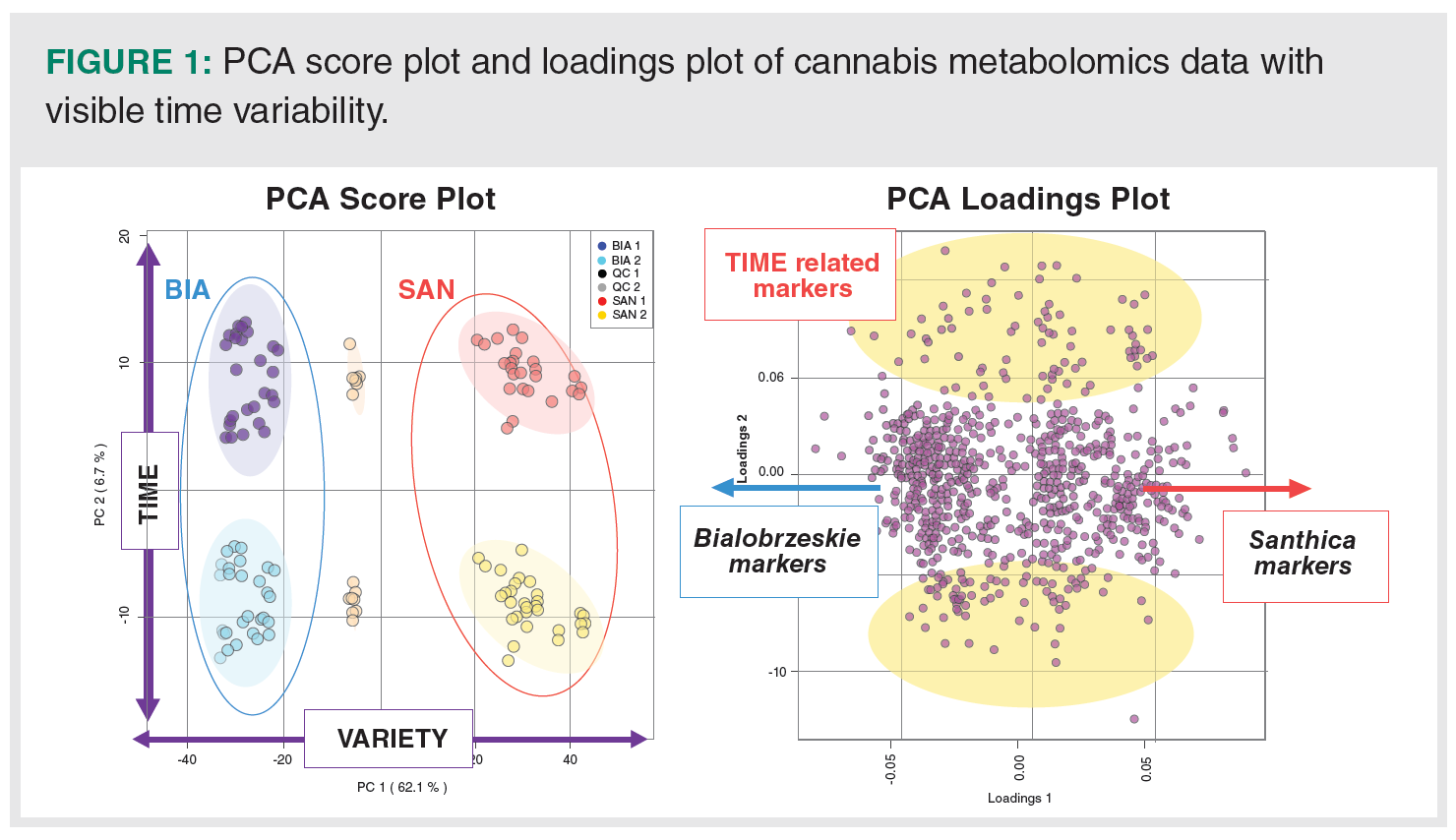
Time Variability in Multivariate Analysis: APCA score plot was used to overview the multivariate data. A standard preprocessing procedure, including TIC normalization, logarithmic transformation, and Pareto scaling, was used (29). Surprisingly, instead of two expected clusters defined by cannabis varieties, four clusters were found (Figure 1). This is very confusing in terms of authentication as it seemed that two other cannabis chemotypes were analyzed. PCA revealed that in addition to the biological variability between Santhica and Bialobrzeskie varieties, which is described by the first principal component and covers 62.1% of the total variance, the instrumental/time variability represents the second largest part of variance (6.7%) in the data. The complementary PCA loadings plot shows the distribution of features. While the points on the first principal component (left and right side) correspond to Bialobrzeskie and Santhica markers, the direction of the second principal component (upward and downward) indicate the increasing effect of time difference on them. In order to calculate the amount of time-related features whose intensity has changed between batches, a paired t-test was performed. There were 486 features significantly differing (FDR, [see Materials and Methods] p-value < 0.01) between QC samples from the first and second batch.


Different Relative Abundance of Features in Time: When investigating the reasons for time-dependent separation, the problem of different relative peak intensities between batches was observed. As the linear range of the time-of-flight mass analyzer used in this study is relatively narrow, the most intensive ions outside of the linearity in the first measured batch could easily get into the linear range in the later date second measurement because of the decreased total ion current. Different peak profiles (caused by peak oversaturation) lead to different peak parameters (area/height), thus the relative abundance of features changes. Figure 2 illustrates the most intensive peak oversaturation in batch 1 and its subsequent fit into linear range in batch 2. As the peak description was inaccurate in batch 1 (the area of the highest peak was significantly decreased by 11%), less abundant peaks were therefore enhanced by 2% and 6%.


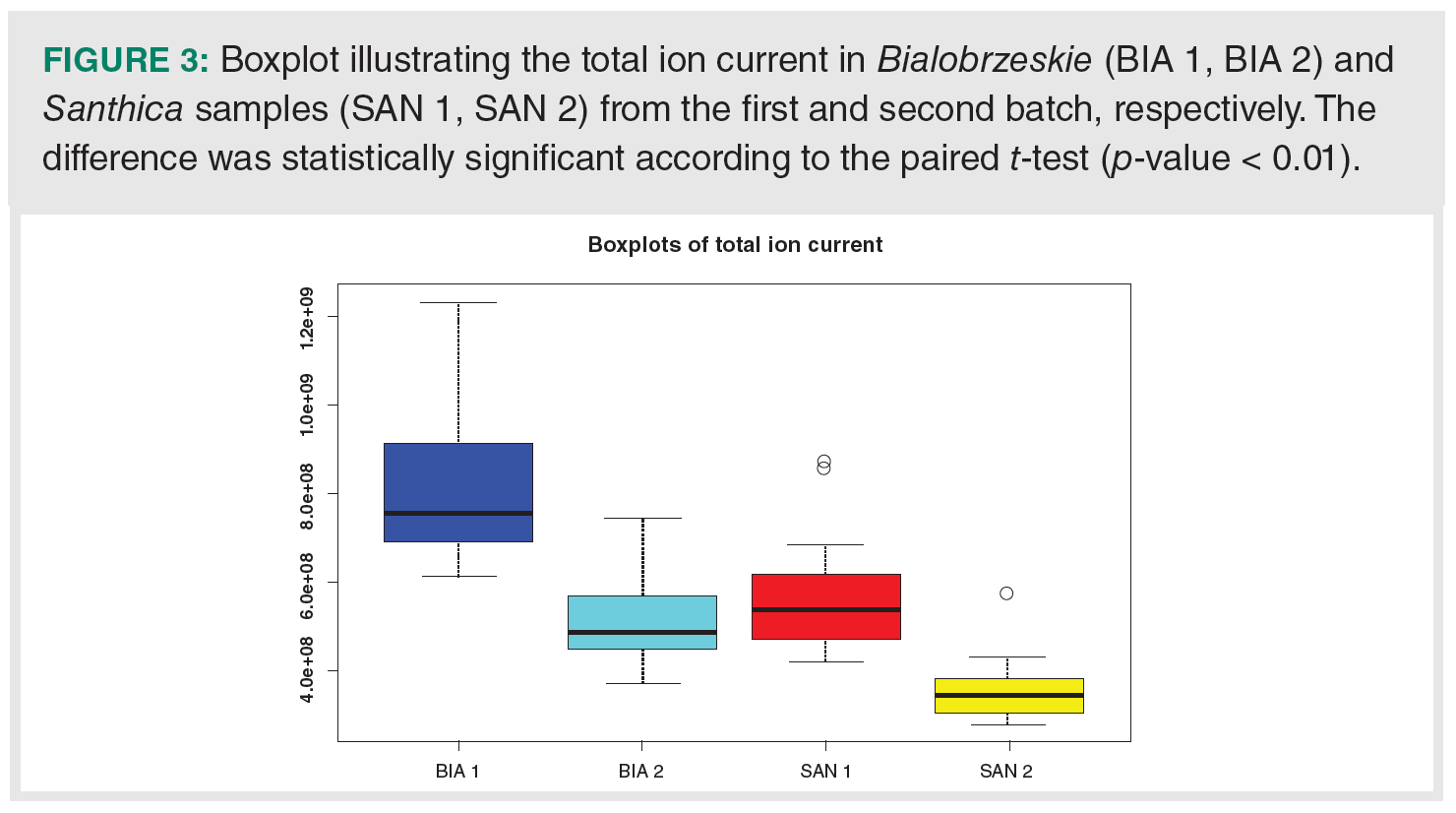
Time dependence could also be observed on the total ion current, which differed not only between cannabis varieties but also with regard to date of analysis. Figure 3 presents the decrease of total ion current in the second batch of the dataset. Based on the paired t-test, the decrease of intensity was significant for both varieties (p-value < 0.01).

Quality Control-Robust LOESS Signal Correction (QC-RLSC) Method: As the connection between the data processing and the following statistical evaluation is very close, the time variability present in the data should be removed. That’s why more advanced normalization techniques were applied. Our developed normalization procedure is based on the QC-RLSC method introduced by Dunn et al. (26). This strategy is based on QC samples that are injected at the same interval throughout the whole sequence. In the case of a larger sample set that cannot be measured in a single run, the same QC sample must be used in all batches. The assumption is still the same sample composition of QC sample through the batch(es) and all changes between individual injections are therefore caused by instrumental variability. In other words, the injection profile serves to monitor these intensity changes. The most pronounced advantage of QC-RLSC is the correction of nonlinear changes, where each variable profile is considered individually. In the first step, only QC sample features intensities are used for locally weighted scatter plot smoothing (LOESS). The fitted curve estimates the values of correction factors for feature intensity in QCs, which are then used for the calculation of correction factors for the rest of the samples using the quadratic spline function. Unlike the QC samples, these analytical samples differ due to the biological variability, and the instrument variability is, depending on the current situation, less or more visible. Their correction factors are therefore estimated based on the QC injection profile. Except for the first and last section of the sequence, the two foregoing and two oncoming QC samples correction factors are always used for polynomial function calculation. The first and last section is resolved with the first and last free QC samples, respectively. Finally, the correction factors for all the samples and features are obtained, and the measured intensities can be divided by them.
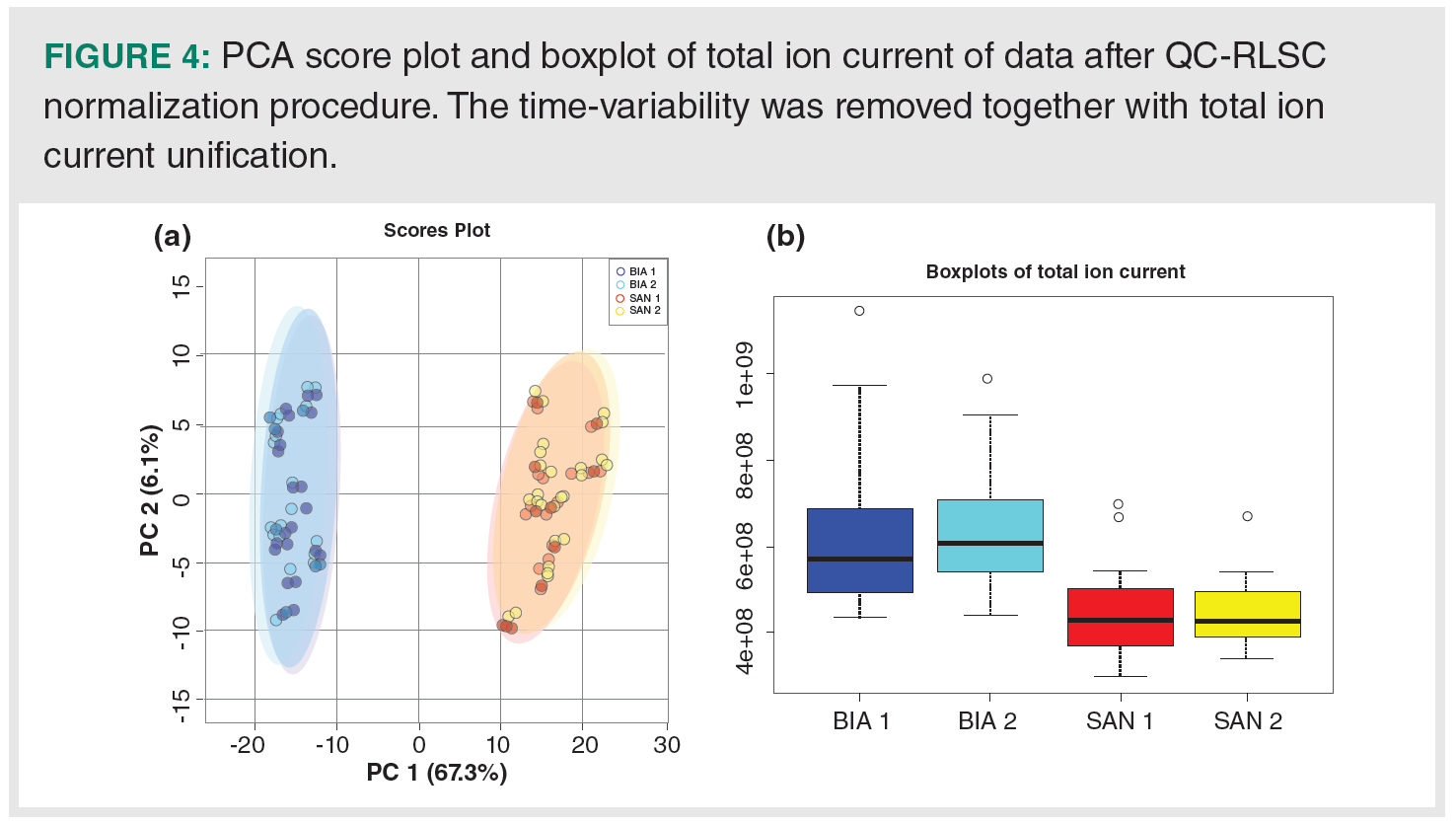
Application of QC-RLSC on Cannabis Fingerprints: After application of QC‑RLSC normalization on the data, the effect of time variability was removed. On the PCA score plot (Figure 4[a]), there is no sign of time variability described by the first two principal components. The total ion current between batches is straightened (Figure 4[b]) and there is also no significant difference between batch 1 and 2 according to the paired t-test, where for Santhica and Bialobrzeskie the p-value was equal to 0.238 and 0.503, respectively. Even when a paired t-test was again used to reveal time‑related markers, none of the features was found to be statistically significantly different (FDR p-value < 0.01) between QC from the first and second batch.

Conclusion
Analytical methods based on UHPLC–HRMS/MS cannot be easily compared, because the metabolomic fingerprint does not have any reference value to which the measured ion intensities could be linked and thus make them time‑dependent. Although the measurement settings were still the same, the condition of the instrument (the status of the ion source and chromatographic system) differ in time and cause the time variability. In initial phase, time dependency was visible after application of the common normalization technique, as exceeding the linear range of the mass analyzer detector occurred. This risk must always be considered. Nevertheless, the correction can be achieved by employing QC-RLSC, a method enabling the removal of undesired time variability by taking each feature intensity profile into account and smoothing it.
Acknowledgement
This study was supported by the project TN01000048 - National Competence Center “Biorefining as a Circulation Technology” co-financed from the Technology Agency of the Czech Republic within the National Competence Center Program.
References
- J.K. Booth and J. Bohlmann, Plant Science 284, 67–72 (2019).
- L.O. Hanuš and Y. Hod, MCA 3(1), 25–60 (2020).
- S.E. Turner, C.M. Williams, L. Iversen, and B.J. Whalley, Phytocannabinoids 103, 61–101 (2017).
- C.M. Andre, J.-F. Hausman, and G. Guerriero, Front. Plant Sci. 7, (2016).
- T. Glivar et al., Industrial Crops and Products 145, 112082 (2020).
- V. Sikora, J. Berenji, and D. Latković, Genetika 43(3), 449–456 (2011).
- T.P. Freeman, C. Hindocha, S.F. Green, and M.A.P. Bloomfield, BMJ 365, l1141 (2019).
- A. Shapira, P. Berman, K. Futoran, O. Guberman, and D. Meiri, Anal. Chem. 91(17), 11425–11432 (2019).
- M.A. ElSohly et al., J. Forensic Sci. 45(1), 24–30 (2000).
- Z. Mehmedic et al., J. Forensic Sci. 55(5), 1209–1217 (2010).
- G. Micalizzi et al., Journal of Chromatography A 1637, 461864 (2021).
- F. Benes et al., LCGC Europe 33(1), 8–16 (2020).
- N. Christinat, M.-C. Savoy, and P. Mottier, Food Chem. 318, 126469 (2020).
- L. Vaclavik et al., J. AOAC Int. 102(6), 1822–1833 (2019).
- K.A. Aliferis and D. Bernard-Perron, Frontiers in Plant Science 11, 554 (2020).
- A. Cerrato et al., Talanta 230, 122313 (2021).
- C. Citti et al., Sci. Rep. 9(1), 20335 (2019).
- M.M. Delgado-Povedano, C. Sánchez-Carnerero Callado, F. Priego-Capote, and C. Ferreiro-Vera, Talanta 208, 120384 (2020).
- M.T. Welling et al., Aust. J. Chem. 74(6), 463–479 (2021).
- Y.H. Choi et al., J. Nat. Prod. 67(6), 953–957 (2004).
- N. Happyana and O. Kayser, Planta Med. 82(13), 1217–1223 (2016).
- A. Hazekamp, K. Tejkalová, and S. Papadimitriou, Cannabis and Cannabinoid Research 1(1), 202–215 (2016).
- B.F. Thomas and M.A. ElSohly, The Analytical Chemistry of Cannabis, 63–81 (2016).
- Q. Meng et al., PLOS ONE 13, e0196396 (2018).
- K.A. Veselkov et al., Anal. Chem. 83(15), 5864–5872 (2011).
- W. Dunn et al., Nature Protocols 6, 1060–1083 (2011).
- J.A. Kirwan, D.I. Broadhurst, R.L. Davidson, and M.R. Viant, Anal. Bioanal. Chem.405(15), 5147–5157 (2013).
- M. Rusilowicz, M. Dickinson, A. Charlton, S. O’Keefe, and J. Wilson, Metabolomics 12, 56 (2016).
- R.A. van den Berg, H.C. Hoefsloot, J.A. Westerhuis, A.K. Smilde, and M.J. van der Werf, BMC Genomics 7(1), 142 (2006).
Kamila Bechynska is a PhD student at the Department of Food Analysis and Nutrition, University of Chemistry and Technology, Prague (UCT Prague), Czech Republic.
Vit Kosek works as a research assistant at the Department of Food Analysis and Nutrition, University of Chemistry and Technology.
Marie Zlechovcova is a research assistant at the Department of Food Analysis and Nutrition, University of Chemistry and Technology.
Petra Peukertova is a PhD student at the Department of Food Analysis and Nutrition, University of Chemistry and Technology.
Jana Hajšlová is a professor at the Department of Food Analysis and Nutrition, University of Chemistry and Technology. She is a head of ISO 17025/2018 accredited laboratory and also heads a research group concerned with separation science in the fields of food, environmental, and metabolomic analysis.











New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Quantifying Terpenes in Hydrodistilled Cannabis sativa Essential Oil with GC-MS
April 21st 2025A recent study conducted at the University of Georgia, (Athens, Georgia) presented a validated method for quantifying 18 terpenes in Cannabis sativa essential oil, extracted via hydrodistillation. The method, utilizing gas chromatography–mass spectrometry (GC–MS) with selected ion monitoring (SIM), includes using internal standards (n-tridecane and octadecane) for accurate analysis, with key validation parameters—such as specificity, accuracy, precision, and detection limits—thoroughly assessed. LCGC International spoke to Noelle Joy of the University of Georgia, corresponding author of this paper discussing the method, about its creation and benefits it offers the analytical community.



