Benefits of Integrating Travelling Wave Ion Mobility Spectrometry into Liquid Chromatography and Mass Spectrometry Workflows for Steroid Analysis
The advantages provided by the implementation of ion mobility spectrometry (IMS), and in particular travelling wave ion mobility spectrometry (TWIMS), in traditional liquid chromatography–mass spectrometry (LC–MS) systems are discussed. TWIMS provides new analytical information on the structure of ions, the so-called collision
cross section (CCS), which in combination with retention indices and mass spectra, increases confidence in compound identification. The use of this parameter for identification purposes is quite recent and the development of CCS databases that can be subsequently applied in both targeted and non-targeted analysis is required.
In the case of steroids, a TWCCSNN2 database for 300 compounds, including endogenous and exogenous steroids, has recently been reported. From the point of view of compound separation, the integration of TWIMS as a third separation dimension in LC–MS workflows increases the resolving power of the method, allowing the separation of isobaric and isomeric steroids. Furthermore, it also allows the separation of the steroids from the chemical background related to the urine sample, improving the limits of detection (LODs) between 2 and 7 times.
Steroid analysis is of special interest in multiple fields, including medicine, food safety, and doping control applications. These biochemical molecules play an important role in human metabolism and are indicators of health and disease (1). Furthermore, the monitoring of steroid levels in professional athletes or animals intended for human consumption is of special relevance to identify their fraudulent use to increase sport performance or as growth promoters in animals, respectively. Traditionally, the analysis of steroids has been approached by gas chromatography–mass spectrometry (GC–MS), although currently the use of liquid chromatography (LC)–MS for this purpose has become widespread (2). Despite the multiple options available to carry out the determination of steroids, it still represents an analytical challenge because of the large number of molecules that make up this chemical family of compounds, their low concentration levels in biological matrices, and the presence of isomers that hamper molecular identification.
The recent commercialization of hyphenated ion mobility-mass spectrometry (IM-MS) instruments offers new possibilities to enhance traditional LC–MS workflows by overcoming the aforementioned challenges. Ion mobility spectrometry (IMS) is a post-ionization separation technique in which ions are separated according to their size, shape, and charge in a gas phase because of the action of an electric field (3). Integration of IMS into LC–MS workflows is possible as separation in the three dimensions occurs on different timescales, namely minutes in LC, milliseconds in IMS, and microseconds in MS. In this sense, a high acquisition rate is required in the MS dimension, so IM-MS instruments are usually based on time-of‑flight (TOF)-MS technology. Regarding IMS, there are several available technologies, each of them presenting advantages and disadvantages with respect to the others (4,5). For non-targeted analysis, which is increasingly applied in holistic studies of the steroidome, three different IMS technologies have been successfully implemented, namely drift tube ion mobility spectrometry (DTIMS), travelling wave ion mobility spectrometry (TWIMS), and trapped ion mobility spectrometry (TIMS).
DTIMS, TWIMS, and TIMS provide information on the collision cross section (CCS) of ions, which represents additional information on the retention indices and mass spectra of the molecules, and which can be used to obtain molecular identification with a higher level of confidence. In IMS, the CCS represents a momentum transfer between the ions and the buffer gas particles in the ion mobility cell, and is derived quantitatively from the mobility of ions (K) via mathematical models such as the Mason-Schamp equation (6). However, this mathematical approach, which is widely used, is only applicable when using DTIMS in a specific operating mode. Consequently, CCS values are generally obtained from calibration curves generated with compounds of known CCS values.
The correlation between CCS and mass-to-charge ratio (m/z) parameters is obvious and undeniable (7). In this framework, there is an open discussion about the advantages provided by the implementation of this molecular characteristic for identification purposes (8). Nevertheless, certain ions have been shown to exhibit unexpected CCS values compared to their chemical and structural analogues (9). It is in these cases where the CCS values are complementary data to m/z, being of special interest for the identification of isobars and isomers that present different CCS under the same experimental conditions. In general, CCS values can be experimentally obtained, computationally generated, or predicted by the use of machine learning approaches (10). Regarding experimentally derived CCS values, an increasing number of CCS databases have been reported in recent years, including a CCS compendium (11,12). In general, early inter-laboratory studies indicate that CCS databases can be applied in laboratories equipped with the same IMS technology as that used for database generation (13,14). However, more research is still needed to evaluate whether extrapolation of CCS values between IM-MS platforms based on different IMS technologies is possible (15).
The use of CCS as an identification parameter in LC–IM-MS-based methods has been shown to reduce the number of false positives/negatives observed in LC–MS analyses (16), but it is not the only advantage provided by IMS. As the movement of ions through the ion mobility cell depends on their conformation in the gas phase, isobars and isomers can be separated depending on the resolving power (Rp) of IMS technology (3). Therefore, this results in a significant improvement in the selectivity of the analytical method. In general, the integration of IMS into LC–MS workflows increases the peak capacity, allowing the resolution of a greater number of ions (5). Moreover, IMS also contributes to the reduction of chemical background noise, improving concentration sensitivity. As a consequence, lower LODs can be reached and compounds can be detected at lower concentration levels in complex matrices (17).
This article covers and groups our most relevant findings related to the analysis of steroids by IMS using TWIMS technology (9,14,17), as well as related computational studies (18). It shows how TWIMS can improve the performance characteristics of LC–MS methods traditionally used for the analysis of steroids, particularly in urine samples. In this sense, it is demonstrated that the integration of TWIMS into LC–MS workflows improves method selectivity and sensitivity, allowing the differentiation of isomeric and isobaric steroids and the detection of steroids at lower concentration levels. Furthermore, the development of a TWCCSNN2 database for steroids, and its subsequent cross-validation by three external laboratories, provides a new tool for steroid identification, especially in non-targeted analysis. In addition, computational studies have been conducted and the application of machine learning tools accessible online has been investigated to provide more confidence in the reported CCS values. Therefore, our work covers the three strategies currently used to generate CCS values. Finally, this article reports the first results obtained for the separation of steroids by cyclic‑TWIMS. They demonstrate how this new commercial technology provides the opportunity to achieve separation of pairs of steroids with close CCS values that could not have been separated by TWIMS before.
Experimental
Chemicals: Water (HiperSolv Chromanorm for HPLC) was supplied by VWR International. Methanol, propan-2-ol, and acetonitrile (LC–MS Chromasolv-grade) were provided by Sigma-Aldrich. Ethanol (Promochem, HPLC-grade) was acquired from LGC Standards GmbH. Formic acid (eluent additive for LC–MS) was purchased from LGC Standards GmbH. Mass calibration was carried out using a solution of sodium formate (0.5 mM in 90:10 (%, v/v) propan-2-ol–water). MS calibration solution was prepared from sodium hydroxide (1 M, Fisher Chemical) and formic acid (Promochem) supplied by Fisher Scientific and LGC Standards, respectively. The Major Mix IMS/TOF Calibration Kit from Waters was used for CCS calibration. Leucine-enkephalin (2 μg/mL) in 50:50 (%, v/v) water–acetonitrile solution containing 0.2% (v/v) of formic acid was used as a lock mass standard. Leucine-enkephalin standard was acquired from Waters. Steroid standards were obtained from Steraloids, Sigma‑Aldrich, and the National Measurement Institute. In general, steroid stock solutions were prepared in ethanol at 100 μg/mLor
1 mg/mL. Working standard solutions (10 μg/mL) were prepared by diluting stock standard solutions with methanol and kept at −20 °C in amber glass vials.
Sample Preparation: Urine samples from bovines (adult animals) and calves were selected from the LABERCA biobank. Urine samples (n = 9) were thawed at room temperature and subsequently prepared following a “dilute-and-shoot” protocol. Briefly, urine samples were submitted to centrifugal filtration for 10 min at 7800 g and 15 °C using centrifugal filters (polyethersulfone membrane, molecular weight cut-off of 10 kDa; VWR International). After filtration, the samples were spiked with working standard solutions consisting of selected mixtures of steroids to achieve a steroid concentration of 2 μg/mL. The spiked samples were subsequently diluted 10-fold with 0.1% (v/v) aqueous formic acid, and submitted to analysis.
Ultrahigh-Performance Liquid Chromatography (UHPLC) Separation: UHPLC separation was performed on an Acquity UPLC system (Waters) equipped with a 2.1 × 100 mm, 1.7‑µm Acquity UPLC BEH C18 column (Waters). Mobile phase consisted of water (A) and acetonitrile (B), and both solvents contained 0.1% (v/v) formic acid. The gradient flow rate was 600 µL/min. The following concentration gradient program was selected for the analysis of steroid standard solutions (except for steroid esters and progestagens) and urine samples: 95/5 (A/B, v/v) between 0 and 0.3 min, 57/43 at 9.6 min, 0/100 from 13.5 to 15.5 min, and 95/5 from 16 to 19.5 min. In the case of steroid ester and progestagen standards, the concentration gradient program consisted of 50/50 (A/B, v/v) between 0 and 2 min, 90/10 at 9.6 min, 0/100 from 13.5 to 15.5 min, and 50/50 from 16 to 19.5 min. The column temperature was kept at 50 °C and 5 µL of samples was injected onto the column.
IM-MS Conditions: IMS analyses were performed on a hybrid quadrupole (Q)-TWIMS-TOF-MS instrument (Synapt G2-S HDMS, Waters) equipped with an electrospray ionization (ESI) interface. Analyses were performed in ESI+ and ESI- modes, acquiring continuous data in the range 150–1200 m/z at 2.5 Hz. The TOF analyzer was operated in high resolution mode. Source and desolvation temperatures were set at 150 and 350 °C, respectively. Nitrogen was used as cone and desolvation gas and was supplied at 50 and 1000 L/h, respectively. Nebulizer pressure was set at 6.0 bar. Cone voltage and source offset were set at 31 and 40 V, respectively. Capillary voltage was set at 3.0 and 2.5 kV for ESI+ and ESI- modes, respectively. A flow rate of 20 μL/min was established for the lock mass standard solution, and MS data were acquired every 15 s at 5 Hz (3 scans to average). LockSpray capillary voltage was set at 3 kV and 2.5 kV for ESI+ and ESI- mode, respectively. A maximum tolerance of 10 ppm was established for identification of ions based on mass accuracy.
Regarding IMS conditions, nitrogen was used as trap and IMS buffer gas at flow rates of 0.2 and 100 mL/min, respectively. The flow rate in the helium cell was set at 180 mL/min. Velocity and height of both StepWave were set at 300 m/s and 5 V, respectively. In the trap cell, wave velocity and height were set at 311 m/s and 4.0 V, respectively. In the transfer cell, wave velocity and height were set at 219 m/s and 4.0 V, respectively. IMS DC bias and trap DC bias were set at 3.0 and 47.0 V, respectively. For analyses in ESI+ mode, IMS wave velocity and height were set at 1000 m/s and 40.0 V, respectively. For analyses in ESI- mode, IMS wave velocity and height were set at 550 m/sand 40.0 V.
TWCCSNN2 Database: For CCS characterization of steroids, steroid standard solutions (10 µg/mL) were injected into the IMS instrument applying a flow injection analysis (FIA) method. The Acquity UPLC System was used for this purpose, although a stainless steel flexible capillary for flow restriction (2 m × 0.12 mm, with 1/16 in female connector on both ends; Agilent Technologies) was selected instead of an LC column. CCS characterization of steroids was performed under IMS conditions similar to those mentioned above. Specific details, including FIA conditions, can be found in the related published paper (9).
Cross-Validation of CCS Values: Cross‑validation of CCS values in the reported TWCCSNN2 database (9) was carried out in three external laboratories located at the University of Geneva (Geneva, Switzerland), Waters Corporation (Wilmslow, UK), and the INRAE-BIA platform BIBS (Nantes, France). These three laboratories are equipped with IMS instrumentation based on TWIMS technology. The INRAE‑BIA platform BIBS was equipped with a Synapt G2-Si instrument (Waters), while Vion instruments (Waters) were available at both the University of Geneva and Waters Corporation. The experimental conditions selected for the cross-validation of the TWCCSN2 database were similar to those LC–IM-MS conditions described above. Details and modifications of these conditions can be found in the related published paper (14).
Cyclic-TWIMS Experiments: Pairs of isomeric steroids were prepared at 62 ng/mL in 1:1 water–acetonitrile containing 0.1% (v/v) formic acid. These stock solutions were infused directly into a Waters cyclic ion-mobility enabled quadrupole time-of-flight (Q-cIM-oaToF) mass spectrometer fitted with an ESI source. The quadrupole was used to select the ions of interest, which were then passed into the cIM region. Scalable IM separation was deployed in a stepwise fashion to observe the arrival time distribution (ATD) profile for zero passes around the cIM device (no IMS separation), followed by one, two, three, and subsequent multiple passes, up to a total of 10 passes. Selection of a narrow portion of ions, followed by fragmentation of those ions, was also carried out.
Computational Studies: The MobCal software was used to calculate the CCS values of the selected steroids whose structures were optimized by density-functional theory (DFT) calculations, using the Gaussian 16 program (19). The ωB97X-D exchange-correlation functional and the Minnesota MN15 functional were used for conformational analysis. The geometry of the various potential ionized structures was fully optimized in vacuo to identify preferred ionization sites. CCS estimations for the various relevant conformers were carried out with the modified version of MobCal incorporating a N2-based trajectory method (TM) algorithm. Finally, CCS values were weighted according to the relevance of the conformers obtained for each steroid.
Results and Discussion
Increase of Confidence in the Identification: CCS parameter has recently emerged as a novel molecular characteristic to support compound identification in combination with retention indices and mass spectra (16). Consequently, a large number of CCS databases are currently being reported in order to overcome the lack of information on this parameter for a wide range of molecular ions (12). In this context, CCS databases should cover the most common and abundant ions found in real sample analysis ([M+H]+, [M+Na]+, [M-H]-, among other relevant ions). However, there are many works in the literature that have addressed the separation of molecular dimers due to their significant differences observed during CCS characterization. From a practical point of view, the analysis of molecular dimers is of little interest since these ions are not always formed during the analysis of complex samples, such as biofluids. Furthermore, if they are formed, detected, and identified on the mass spectrum, they are usually present in much lower abundance than other monomer ions observed for the same molecule. Therefore, to extend the use of IMS beyond the field of academic research, CCS characterization, as well as ion separation by IMS, must focus on those ions that are likely to be detected in the analysis of real samples.
TWCCSNN2 Database for Steroids: The CCS database for steroids includes up to 300 compounds, such as protonated and deprotonated molecules and sodium adducts, among many others, and covers endogenous compounds, such as glucuronide and sulfate metabolites, and exogenous substances, such as steroid esters, as well as deuterated steroids (9). It is the first large CCS database reported for this family of compounds and CCS values have been obtained using nitrogen as buffer gas in a TWIMS‑TOF‑MS system. Therefore, following currently accepted nomenclature (6), this database is referred as TWCCSNN2 database for steroids. It is important to indicate the type of IMS technology and buffer gas used for CCS characterization, since the CCS of compounds depends on experimental conditions, such the type of gas in the IMS cell, temperature, and pressure. Furthermore, it is currently not demonstrated that CCS databases can be extrapolated and applied in instruments based on different IMS technologies.
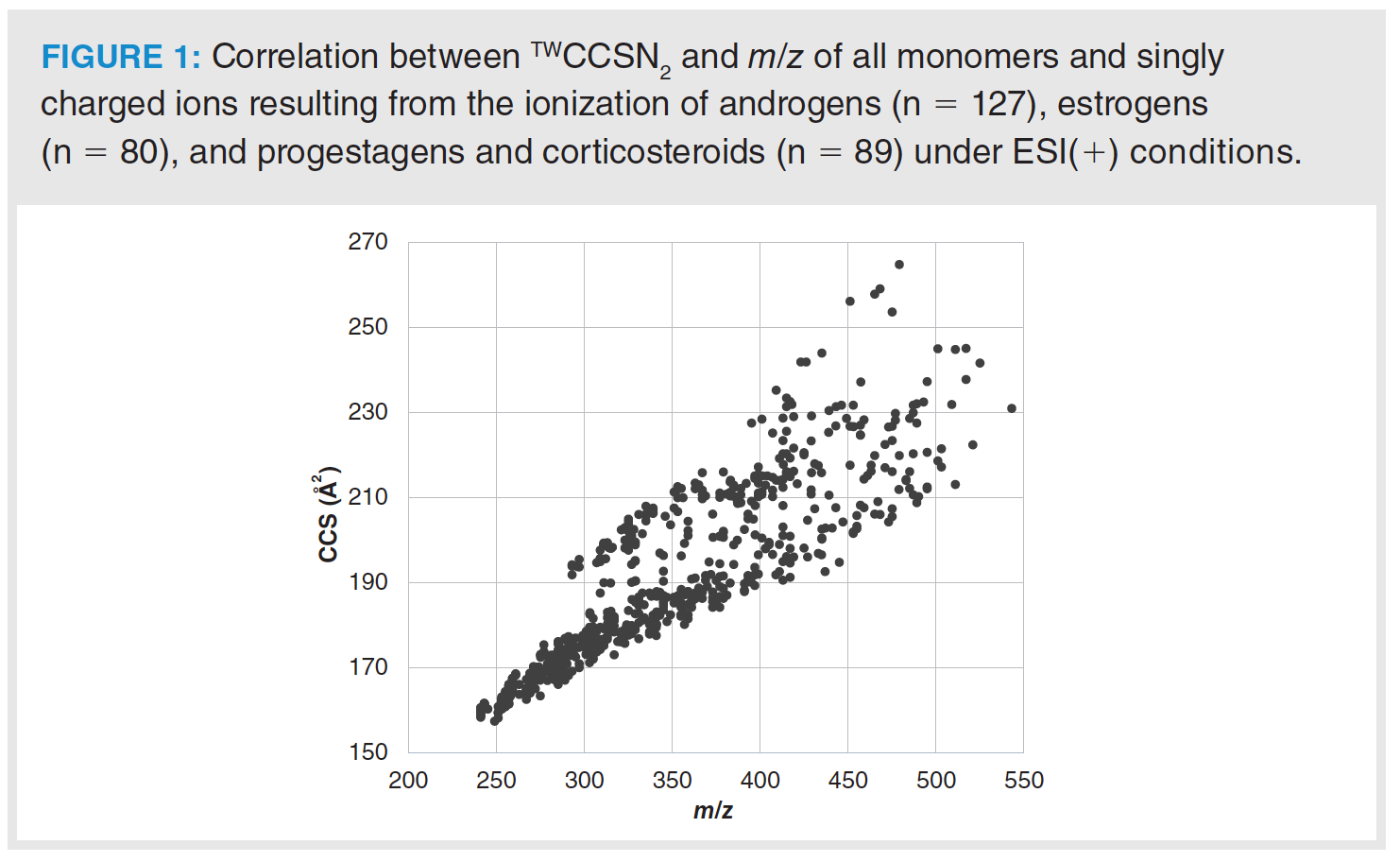
The TWCCSNN2 database for steroids covers a m/z range between 241.1956 and 543.2126, while TWCCSNN2 ranges from 157.3 to 264.8 Å2 if only monomers and singly charged ions are selected. Figure 1 shows the relationship between TWCCSNN2 and m/z observed for positively charged steroid ions. Although both parameters cannot be considered orthogonal, the observed CCS differences for ions with identical or close accurate mass are sufficient to distinguish these ions. For example, the sodium adduct of 6β-hydroxyetiocholanolone (m/z 329.2087) exhibits a totally different TWCCSNN2 (190.3 Å2) from the TWCCSNN2 observed for the positional isomers 11β-hydroxyetiocholanolone and 16α-hydroxyetiocholanolone (m/z 329.2087; 199.3 Å2 and 195.1 Å2, respectively). The TWCCSNN2 of sodium adducts also differs greatly in the case of other steroids, such as 18-hydroxycortisol (m/z 401.1935; 200.4 Å2) and 6β-hydroxycortisol (m/z 401.1935; 211.4 Å2). These examples show how IMS can be a powerful parameter to contribute in compound annotation in non-targeted analysis or to improve confidence in identifying compounds in targeted analysis, especially to distinguish between isomers.


Cross-Validation of CCS Values: Cross‑validation of the reported CCS databases seems necessary to extend their use beyond the laboratories where they have been generated. As mentioned earlier, CCS values can be obtained experimentally, computationally calculated, or predicted using machine learning approaches (10). Initially, the TWCCSNN2 database for steroids was cross-validated experimentally by three external platforms using TWIMS technology, specifically in a Synapt G2-Si and two Vion instruments (14). For this purpose, 97 steroids included in the original TWCCSNN2 database were selected. The selection encompassed compounds from the different classes of steroids, such as androgens, n = 42; estrogens, n = 28; progestogens, n = 8; and corticosteroids, n = 19, and involved a maximum of 167 ions under study. In general, the TWCCSNN2 measurements performed on the Synapt G2-Si, Vion #1, and Vion #2 instruments were biased within the ±2.0% range of the TWCCSNN2 values in the database for 98.8, 79.9, and 94.0% of the ions detected by each platform (a minimum of 142 ions), respectively. This bias is within the expected bias threshold, as a threshold of ±2.0% is widely accepted for TWCCSNN2 measurements compared to TWCCSNN2 values in databases. Therefore, the TWCCSNN2 database for steroids has been successfully cross-validated. Nevertheless, and in order to reduce the bias between TWCCSNN2 values measured in other external laboratories and reference values, a TWCCSNN2 database with average TWCCSNN2 values from the four different laboratories (LABERCA, University of Geneva, Waters Corporation, and the INRAE-BIA) has been reported (14). In this database, the relative standard deviations (RSDs; n = 12) were below 1.5%. The bias between the average TWCCSNN2 values (or reference values) and the TWCCSNN2 measurements performed by each laboratory was within the range of ±1.5% for 96.8% of the total ions (n = 142).
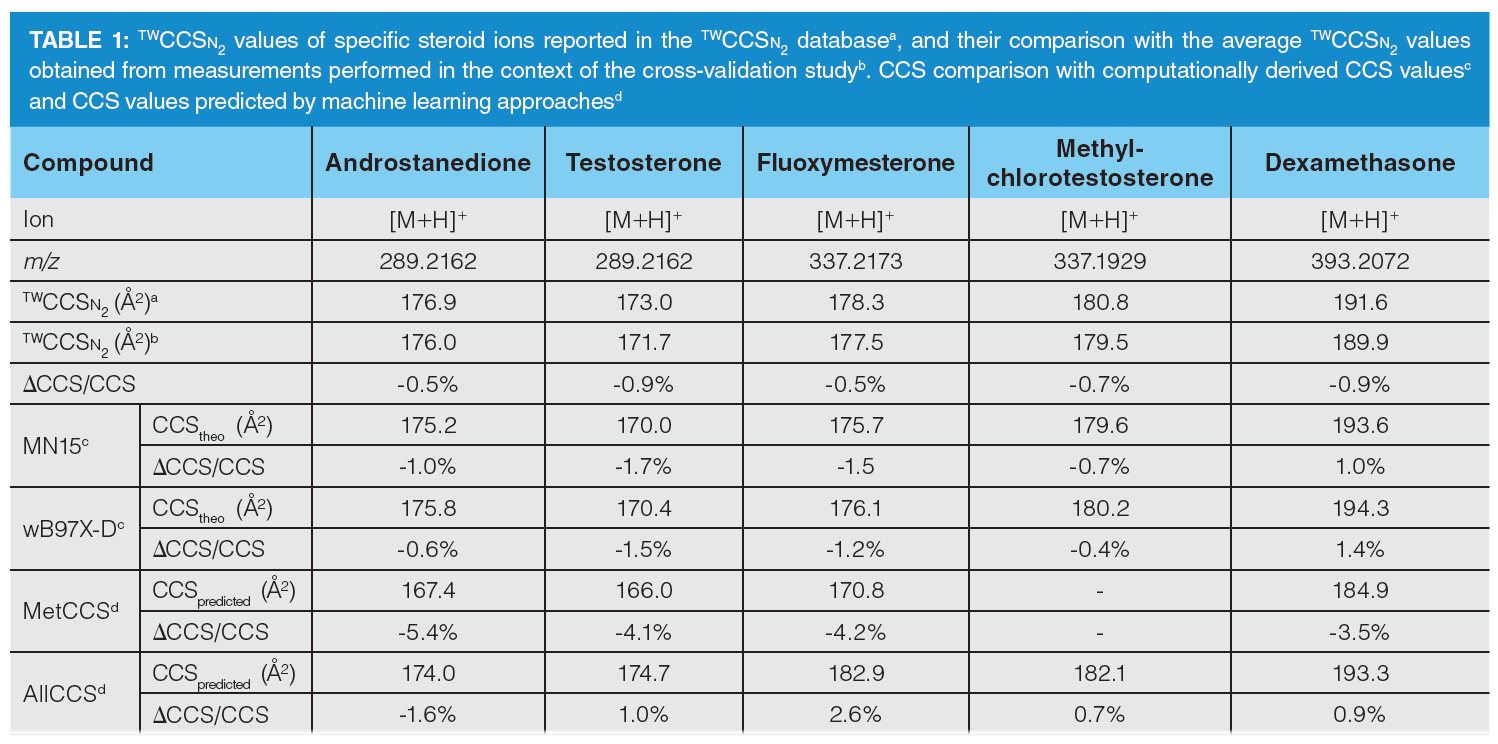
Computational studies and the application of machine learning approaches were carried out for a smaller number of steroids to verify the potential of both methodologies to generate CCS values and, consequently, be used as tools for cross-validation of the reported CCS databases. First, computationally-derived values were obtained for the selected 20 steroids (involving 23 ions) applying two different approaches (18), as specified in the experimental section. In this sense, slightly better performance was observed for the MN15 functional in comparison to that of the ωB97X-D functional. In general, high correlation was observed between experimental (9) and computationally‑derived CCS values (R2= 0.9696 for MN15 functional). Furthermore, the CCS value for almost 80% of the studied compounds was properly calculated within a ±2% error range from the observed data. Therefore, although time-consuming, computational studies are suitable for cross-validation of CCS databases, as well as generating information on the CCS of compounds that are not available as chemical standards and cannot be characterized. Finally, two machine learning tools, MetCCS (20) and AllCCS (21), which are accessible online, were evaluated for the prediction of steroid CCS values. Table 1 shows the CCS values of a selection of five steroids obtained by the three different approaches currently available to generate CCS values, including by machine learning tools. Although machine learning approaches are useful tools for rapidly generating CCS values compared to time‑consuming computational approaches, they cannot be as precise regarding experimentally measured CCS values. Obviously, as can be seen from Table 1, the precision of machine learning approaches depends on the model itself, such as the parameters used to build the model, as well as the compound dataset used to generate the model.

Matrix Effect: In order to implement the use of the CCS values as an identification parameter, it seems important to demonstrate that there is no influence of the sample matrix on the CCS measurement of the CCS of the analyte of interest. Such effect matrix was evaluated for phase II metabolites of androgens and estrogens (n = 25) in urine samples, studying both positive and negative ionization mode (17). All steroids were detected as the deprotonated ion under ESI(-) conditions, whereas only 21 steroids were detected either as the protonated molecule or as a sodium adduct under ESI(+) conditions. In total, 1086 TWCCSN2 measurements were carried out over four months, and high accuracy was observed for TWCCSN2 values of steroids in urine samples compared to the TWCCSN2 values included in the database. More than 77% of TWCCSNN2 measurements for steroids in urine samples were within an error of ±0.5%, while this error was only greater than ±1.0% in 1.4% of the TWCCSNN2 measurements. Therefore, the urine matrix did not show any influence on the TWCCSNN2 measurement of steroids, since the TWCCSNN2 values obtained in the presence of matrix matched with the TWCCSNN2 values in the database within the extensively applied error threshold of ±2.0% for the comparison of measured TWCCS values and TWCCS values in databases (22).
Selectivity Improvement: The low Rp of TWIMS technology available as part of IM-MS instruments has traditionally been a drawback to achieving the separation of isomers and isobars showing close CCS values. In some ways, this fact has probably been the main limitation for the development of applications using this analytical technique. For compounds with CCS values around 200 Å2, only those compounds that show a difference in terms of CCS of at least 2.5% can be separated in a TWIMS dimension with a Rp of 40 (23). However, the introduction of cyclic-TWIMS instrumentation has brought the possibility of separating with TWIMS technology those complex mixtures of isomeric and isobaric compounds that present smaller differences.
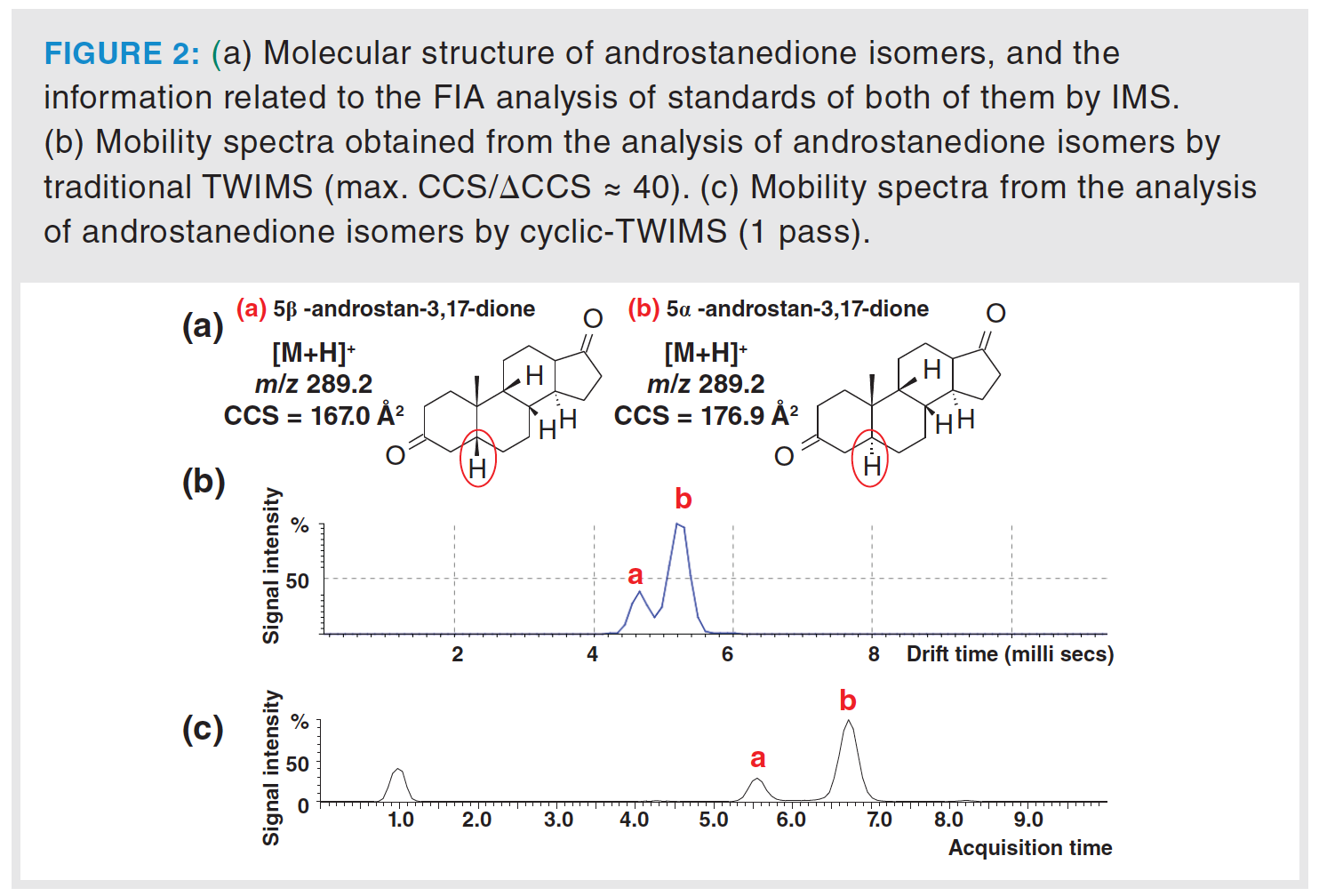
In the case of steroids in urine samples, we have observed that only those ions that differ in CCS values by at least 4%, such as the deprotonated molecules of etiocholanolone glucuronide (TWCCSNN2 = 206.9 Å2) and epiandrosterone glucuronide (TWCCSNN2 = 221.4 Å2) or 19-noretiocholanolone glucuronide (TWCCSNN2 = 204.2 Å2) and 19-norandrosterone glucuronide (TWCCSNN2 = 213 Å2), can be separated by the Synapt G2-S system. Even in spite of that, partial or no separation was observed for other steroid isomers, even when they showed a CCS difference greater than 4%. For example, androstanedione isomers, which differ in the α or β position of the proton at C5 and show a CCS difference of 5.8% for their protonated forms (Figure 2[a]), were only partially separated in the TWIMS dimension of the Synapt G2-S instrument (Figure 2[b]). However, these ions have been separated at the baseline by a cyclic-TWIMS system, even passing the ions only once through the ion mobility cell (Figure 2[c]). In this sense, the Rp of the cyclic-TWIMS selecting a single pass is already greater than that of the Synapt instruments (Rp of 65 vs. 40) (24).

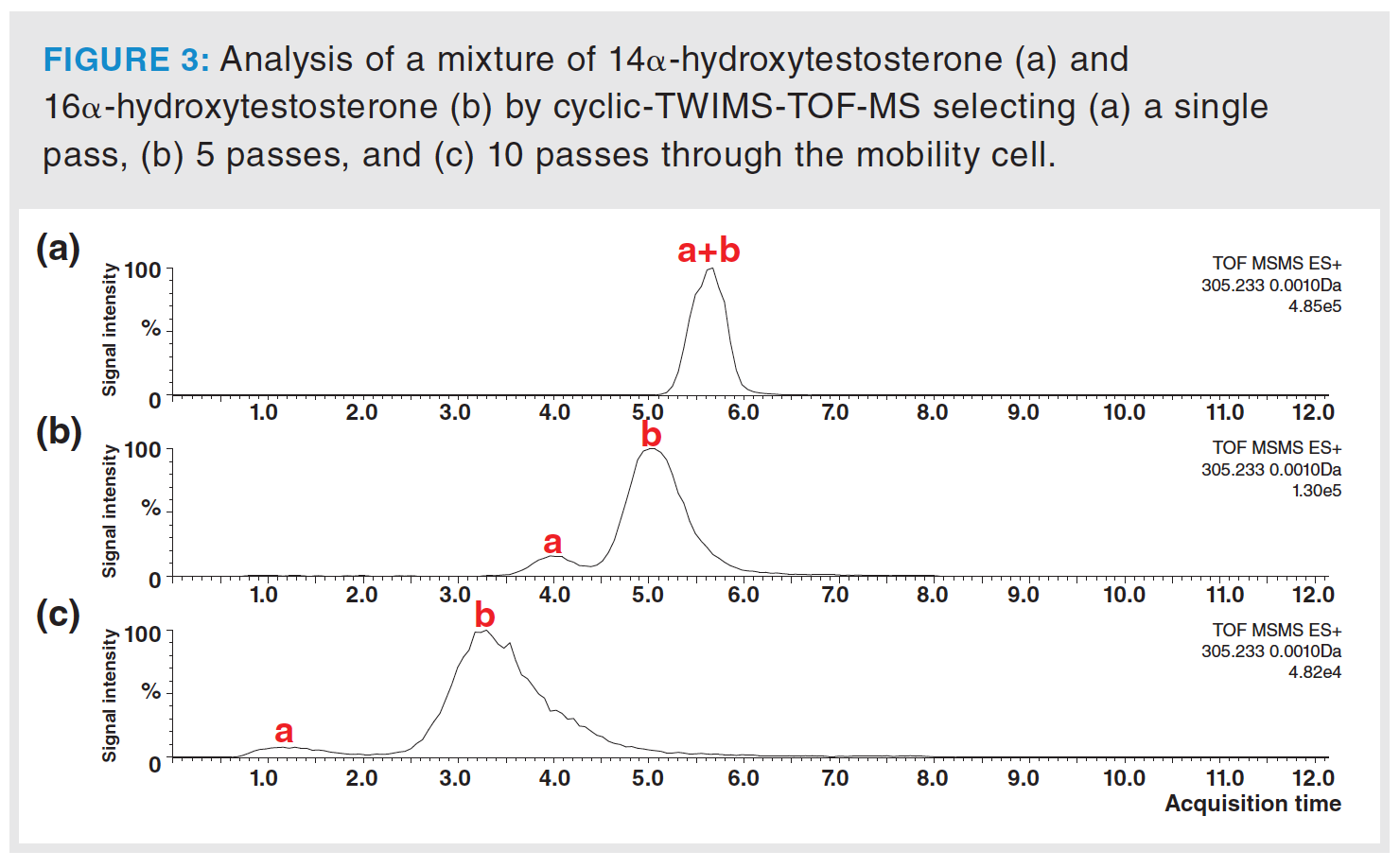
Other isomeric pairs of steroids, such as 14α-hydroxytestosterone and 16α-hydroxytestosterone, did not separate on either the Synapt G2-S system or the cyclic-TWIMS instrument by selecting only a single pass. In fact, the protonated ions of both molecules (m/z 305.2111) show close TWCCSNN2 values, 174.2 and 176.1 Å2, respectively, requiring a high resolving power of the TWIMS dimension to be separated. Since the resolving power of the cyclic-TWIMS system depends on the number of passes selected, up to 10 passes were evaluated to achieve baseline separation of these ions with a CCS difference of 1.1%. Figure 3 shows how both ions practically separate at the baseline when they pass through the cyclic-TWIMS path five times. In this sense, it could be argued that the separation would improve if they were made to pass a greater number of times through the mobility cell. However, this is not without its drawbacks, as ions cannot be passed through the cyclic-TWIMS cell indefinitely. In addition to a significant loss of ions in the successive passes through the cell, the increasingly narrow selection of the range of mobility to separate in each pass leads to obtaining broader peaks and poorer quality analytical information. As can be seen in Figure 3(c), a significant depletion of signal intensity is observed when ions move along the cyclic-TWIMS path ten times instead of five times; broader peaks can also be observed. Therefore, the number of passes to be selected in cyclic‑TWIMS implies a compromise between the resolving power to be achieved and signal intensity and/or peak shape.

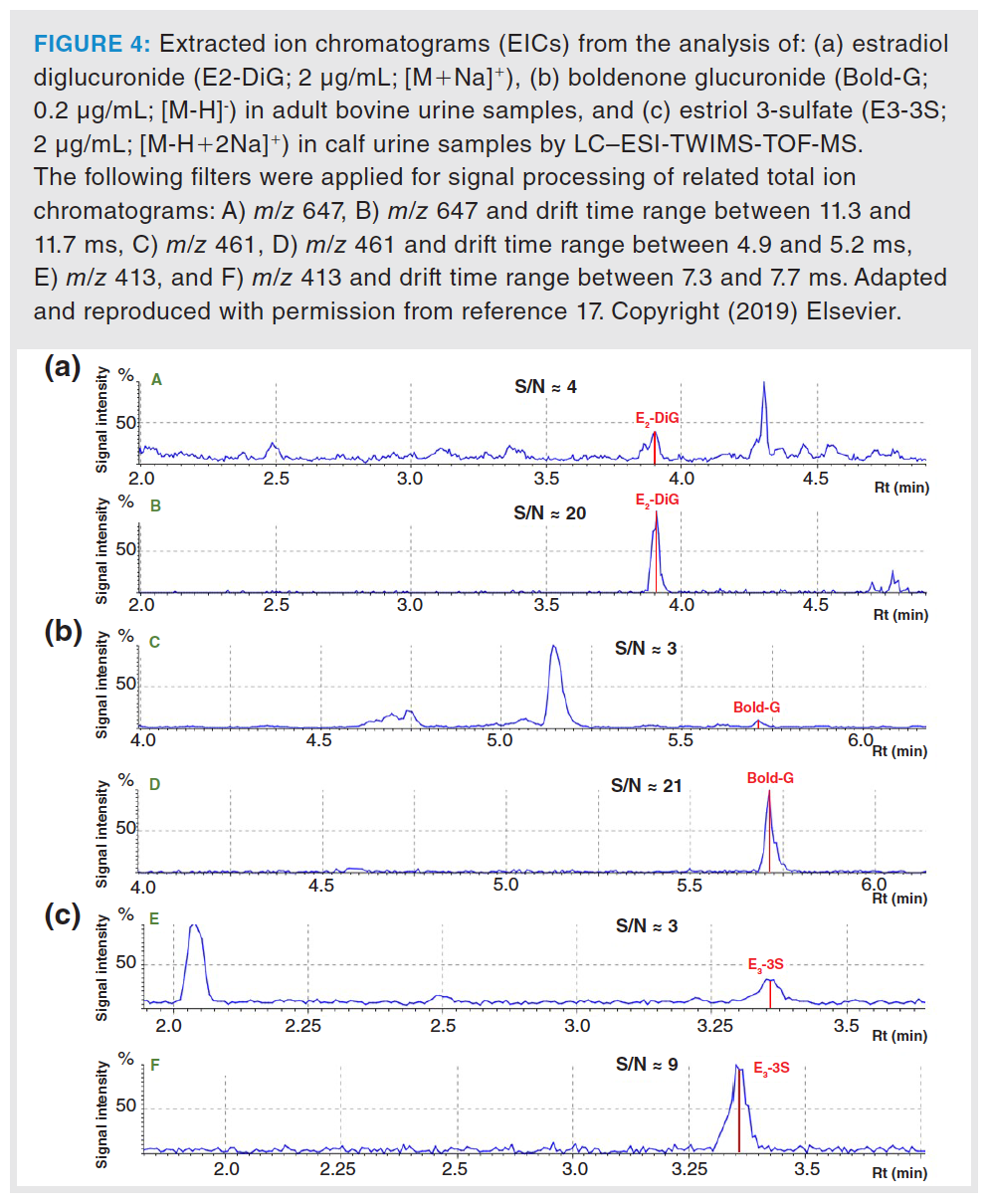
Sensitivity Enhancement: One of the main advantages of integrating IMS into LC–MS workflows is that it generally improves method sensitivity. This third dimension brings the possibility of selecting drift time regions (or CCS values) of interest in the acquired data, which implies the subtraction of the analytical signal of the analytes from the chemical background noise. Consequently, lower LODs can be achieved. Figure 4 shows the improvement in terms of LODs observed for various phase II steroid metabolites when the same urine samples were analyzed by LC–TWIMS-MS and LC–MS (TWIMS cell was inactivated but allowed ion transmission). In general, signal-to-noise ratio (S/N) was improved from two- to sevenfold by selecting the drift time region of the target analytes. As can be seen in Figure 4, this improvement in concentration sensitivity is crucial for the determination of compounds at concentration levels close to the LOD of the method (S/N = 3). They can be determined at concentration levels even higher than the limit of quantification (LOQ) (S/N = 10) when including the IMS as a third separation dimension, therefore, allowing their quantification.

Conclusions
TWIMS or other IMS technologies, such as DTIMS or TIMS, are called to revolutionize current LC–MS methods, or other methods that involve chromatographic separations and MS detection. As observed for this particular case of steroids, it introduces an extra separation dimension in LC–MS workflows that enhances concentration sensitivity and increases the resolving power of the method. Furthermore, the molecular ions of isomers, such as those of steroids, can be distinguished according to their TWCCSN2. Isomer differentiation has been a traditional analytical challenge because they tend to show similar mass spectra and can coelute in the chromatographic dimension. As shown in this work, IMS is a powerful analytical technique to overcome that challenge.
Although low resolving power has traditionally been attributed to IMS-based systems, in recent years, technological improvements have been made regarding this drawback. Consequently, there are IM-MS instruments currently available that allow the separation of compounds showing similar ion mobility or close structural conformation in the gas phase. Therefore, ions with close CCS values are now susceptible to being separated by IMS. In this sense, in the coming years a greater implementation of the use of the CCS in both targeted and non-targeted approaches is expected with the aim of improving confidence in the identification of compounds. However, it is still necessary to characterize a wide range of molecules in terms of CCS to generate large CCS databases that can be applied routinely in compound determination. It is desirable that such CCS databases are based on measurements carried out in different laboratories, thus reducing any laboratory bias. Also, computational calculations or even proper machine learning tools are suitable approaches for generating CCS values and expanding the number of entries in CCS databases, especially for those compounds for which analytical standards are not available.
Acknowledgements
We thank the following researchers for their contributions to the generation of part of the results included in this article: Fabrice Monteau (LABERCA; Nantes, France); Valentina D’Atri, Julian Pezzatti, Serge Rudaz, and Davy Guillarme (University of Geneva; Geneva, Switzerland); Mathieu Fanuel, David Ropartz, and Hélène Rogniaux (INRAE-BIA platform BIBS; Nantes, France); Gitte Barknowitz, Nicola Dreolin, Eleanor Riches, and Sara Stead (Waters Corporation; Wilmslow, UK); Samuel Normand, Jérôme Graton, and Jean-Yves Le Questel (Université de Nantes; Nantes, France).
References
- E. Olesti, J. Boccard, G. Visconti, et al., J. Steroid Bichem. Mol. Biol. 206, 105797 (2021).
- C. Gomez, A. Fabregat, Ó.J. Pozo, et al., TrAC Trend Anal. Chem. 53, 106–116 (2014).
- V. D’Atri, T. Causon, O. Hernandez-Alba, et al., J. Sep. Sci. 41, 20–67 (2018).
- F. Lanucara, S.W. Holman, C.J. Gray, et al., Nat. Chem. 6, 281–294 (2014).
- G. Paglia, A.J. Smith, and G. Astarita, Mass Spec. Rev. DOI:10.1002/mas.21686 (2021).
- V. Gabelica, A.A. Shvartsburg, C. Afonso, et al., Mass Spec. Rev. 38, 291–320 (2019).
- M.-D. Luo, Z.-W. Zhou, and Z.-J. Zhu, Drug Test. Anal. 4, 163–174 (2020).
- A. Kaufmann, P. Butcher, K. Maden, et al., Anal. Chim. Acta 1107, 113–126 (2020).
- M. Hernández-Mesa, B. Le Bizec, F. Monteau, et al., Anal. Chem. 90, 4616–4625 (2018).
- Z. Zhou, J. Tu, and Z.-J. Zhu, Curr. Opin. Chem. Biol. 42, 34–41 (2018).
- K.M. Hines, D.H. Ross, K.L. Davidson, et al., Anal. Chem. 89, 9023–9030 (2017).
- J.A. Picache, B.S. Rose, A. Balinski, et al., Chem. Sci. 10, 983-993 (2019).
- S.M. Stow, T.J. Causon, X. Zheng, et al., Anal. Chem. 89, 9048–9055 (2017).
- M. Hernández-Mesa, V. D’Atri, G. Barknowitz, et al., Anal. Chem. 92, 5013–5022 (2020).
- V. Hinnenkamp, J. Klein, S.W. Meckelmann, et al., Anal. Chem. 90, 12042–12050 (2018).
- S. Goscinny, M. McCullagh, J. Far, et al., Rapid Commun. Mass Spectrom. 33, 34–48 (2019).
- M. Hernández-Mesa, F. Monteau, B. Le Bizec, et al., Anal. Chim. Acta: X 1, 100006 (2019).
- J. Graton, M. Hernández-Mesa, S. Normand, et al., Anal. Chem. 92, 6034–6042 (2020).
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, (2016).
- MetCCS, machine learning tool. http://www.metabolomics-shanghai.org/MetCCS/prediction/. Accessed 21 Sep 2021.
- AllCCS, machine learning tool. http://allccs.zhulab.cn/. Accessed 21 Sep 2021.
- M. Hernández-Mesa, D. Ropartz, A.M. García-Campaña, et al., Molecules 24, 2706 (2019).
- J.N. Dodds, J.C. May, and J.A. McLean, Anal. Chem. 89, 12176–12184 (2017).
- J. Ujma, D. Ropartz, K. Giles, et al., Anal. Chem. 30, 1028–1037 (2019).
Maykel Hernández-Mesa is a postdoctoral researcher evaluating the potential of IM-MS for steroidomics in the context of food safety.
Gaud Dervilly is senior researcher, head deputy of LABERCA joint research unit (Oniris/Inrae), Nantes.
Bruno Le Bizec is a professor in food safety at the National Veterinary College of Nantes, Oniris. He is the Head of LABERCA.













Evaluating Body Odor Sampling Phases Prior to Analysis
April 23rd 2025Researchers leveraged the advantages of thermodesorption, followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC×GC/TOF-MS), to compare and assess a variety of sampling phases for body odor.



