Boosting the Purification Process of Biopharmaceuticals by Means of Continuous Chromatography
Special Issues
Single-column (batch) chromatography, involving two or more successive single-column (batch) chromatographic steps, is a standard approach for purifying biopharmaceuticals. Step one, known as the capture step, is used to remove product-related impurities, and step two, the polishing step, is used to remove product-related impurities. Here we present and illustrate the advantages of continuous chromatography for these separations: capture simulated moving bed (captureSMB) for the capture step and multicolumn countercurrent solvent gradient purification (MCSGP) for polishing.
Many biopharmaceuticals are currently purified by means of two or more successive single-column (batch) chromatographic steps. The first one is usually a capture step, which is used to remove non-product-related impurities, such as host-cell proteins and DNA. The second step is referred to as the polishing step, which removes product-related impurities, such as fragments and aggregates. However, single-column processes suffer some intrinsic limitations. Indeed, in the capture step, the trade-off between capacity utilization and productivity can be very relevant, while polishing processes are characterized by yield-purity trade-off. These limitations can be alleviated through continuous, or semi-continuous, countercurrent purification techniques. These processes display superior purification performance, allowing for the automated internal product recycling in the system composed of multiple identical columns, either interconnected or operated in parallel. In this paper, the advantages of capture simulated moving bed (captureSMB) for the capture step and multicolumn countercurrent solvent gradient purification (MCSGP) for polishing purposes will be illustrated.
Biopharmaceuticals have rapidly grown in popularity among the medical community in recent years, as a result of unprecedented advancements in biologics and human genetics. Due to their high affinity toward a specific molecule or receptor, biomolecule-based therapeutics have been proven to have very high efficacy even at low concentrations. Moreover, endogenous (or endogenous-like) biomolecules are better tolerated by human bodies than traditional therapeutics, preventing or diminishing the occurrence of side effects after their administration. For these reasons, biological drugs for the treatment of already existing and emerging diseases represent the basis for tomorrow’s medicine.
The sudden outbreak of the COVID-19 pandemic disease caused by the new coronavirus 2019-nCoV (now oï¬cially designated as severe acute respiratory syndrome-related coronavirus, SARS-CoV-2), has led to an urgent demand for novel therapies for the treatment of clinically advanced conditions. Several options can be taken into consideration for the treatment or prevention of COVID-19, mostly based on the use of biopharmaceuticals, including vaccines, monoclonal antibodies (mAbs), oligonucleotide-based therapies, peptides, interferon therapies and small-molecule drugs (1–4). Particularly relevant is the case of the mAb tocilizumab, under clinical evaluation for its ability to prevent the inflammatory process responsible for the worsening of pneumonia and pulmonary distress in patients affected by COVID-19 (4).
The industrial production of biopharmaceuticals has rapidly progressed in the last few years. However, the recent developments in cell culture and fermentation processes (such as for the production of mAbs) and solid-phase synthesis (for the production of peptides and oligonucleotides, for example) have not been matched by equivalent improvements in purification (downstream) processes, which often represent the bottleneck, in terms of both cost and time, in the entire production process (5).
Preparative liquid chromatography is the preferred method of choice to achieve the purified target at an acceptable degree of purity for therapeutics (6,7). Most of the modern downstream processes need at least two single-column purifications. The first one is usually called capture step, which serves to remove all non-product-related impurities, such as host-cell proteins and DNA. Successive polishing steps are then used to obtain the target at the desired degree of purity, by removing all product-related impurities. These are species, produced during the synthesis, with very similar chemical characteristics to the target compound (such as, truncated or deamidated species and aggregates, for example). The removal of these impurities via chromatography is very challenging, because their chromatographic behavior is often similar to that of the target. This situation very often leads to overlapping regions in the chromatogram where target and impurities are coeluted. The collection of these regions improves the yield of the separation at the expense of the overall purity. On the other hand, the discharge of these regions saves the overall purity at expenses of the process yield. These considerations are at the basis of the well-known purity-yield trade-off, affecting the performance of elution chromatography.
Among the strategies that can enhance the downstream process, multicolumn countercurrent continuous, or semi-continuous, chromatographic techniques seem to be particularly suitable. One of the greatest advantages of continuous techniques is that the purification process can be completely automated, with no human intervention, with a considerable saving of time. These approaches involve the use of two (or more) “identical” columns of the same dimensions and stationary phase, connected through a series of valves. This system allows not only the internal product recycling of the overlapping regions for enhanced product–impurity separation, but also to simulate the apparent opposite movement of the stationary phase with respect to the mobile one, from where the term countercurrent is derived to refer to these techniques.
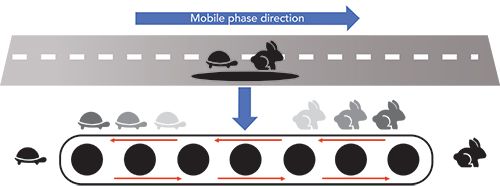
The countercurrent separation of two compounds can be explained through the simple graphic represented in Figure 1. Let us imagine that a slower turtle and a faster rabbit are moving in the direction of the blue arrow (right). Suddenly, they fall onto a conveyor belt moving in the opposite direction (left). Depending on the relative velocities of the turtle and the rabbit (compared to that of conveyor belt that can be properly varied), the slow turtle will be transported to the left of the conveyor belt, while the fast rabbit will continue its run to the right. At the end, the two animals will be separated at the opposite sides of the conveyor belt. In this representation, the turtle is the strongly adsorbed compound (slower velocity into the column), while the rabbit is the weakly adsorbed one (moving faster). The blue arrow represents the direction of the mobile phase. Finally, the conveyor belt represents the countercurrent movement of the stationary phase.


Figure 1: Schematic representation of the countercurrent mechanism; see text for details. Shadowed images of the turtle and the rabbit serve to simulate their movements. Modified with permission from reference (7).
The first countercurrent multicolumns setup was simulated moving bed (SMB) applied for the first time more than 60 years ago for the separation of binary mixtures (8–11). Since then, the SMB concept has been modified and improved, particularly in the direction of reducing the number of columns connected together. This paper focuses, in particular, on two of the most recent improved versions of the traditional SMB concept, captureSMB and multicolumn countercurrent solvent gradient purification (MCSGP). Their advantages over traditional single-column techniques for the purification of therapeutic biomolecules are illustrated.
CaptureSMB
The capture step usually deals with very large volumes of feed coming from the upstream process containing a large number of non-product-related impurities. An affinity resin is used to selectively capture the target molecule. All the other impurities will not bind to the stationary phase, and, therefore, they can be easily removed.
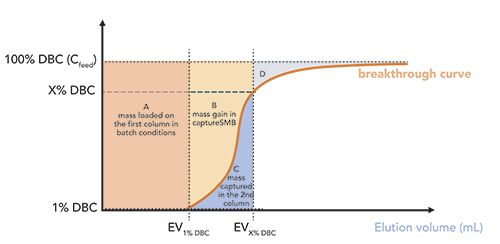
Let us consider a typical case where capture processes are employed-the purification of mAbs with Protein A stationary phase (12). In batch chromatography, the feed is injected into the column by adjusting the loading on the base of the dynamic binding capacity (DBC) value, which can be experimentally evaluated by a breakthrough curve (see Figure 2). A 1% DBC (the capacity at 1% of the breakthrough curve) is taken as reference limit to indicate the saturation of all available affinity sites on the stationary phase. By loading the column beyond this limit, there would be a loss of the target, which would not bind to the stationary phase. Therefore, in batch processes, the column is usually loaded up to 80–90% of 1% DBC, with a 10–20% margin in order to avoid any target-compound loss. After the loading, the target is eluted from the column and the resin is washed and regenerated.

Figure 2: Schematic representation of a breakthrough curve. Area A represents the mass that can be loaded in the first column in batch conditions to reach 1% DBC. In twin-column capture SMB, the mass loaded on the first column is given by A + B while mass C is captured on the second column. The maximum saturation capacity of the stationary phase is given by the sum of masses A + B + D. EV1% DBC and EVX% DBC are the elution volumes at 1% and X% DBC, respectively.
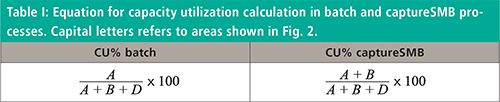
Even if very high yield and purity can be obtained by means of batch purifications, there is an intrinsic trade-off between capacity utilization and productivity. Capacity utilization (CU) is defined as the ratio between the loading (L) and the maximum saturation capacity of the stationary phase (Qsat), which also corresponds to the static binding capacity (SBC):

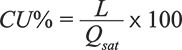
Productivity (for an n-column process) is defined as:

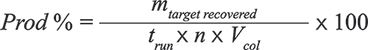
where mtarget recovered is the mass of the target collected at the end of the run, trun is the duration of a run and Vcol is the geometrical volume of the column. For a batch process, n = 1. Productivity is expressed in g/L/h.
To explain the trade-off of batch capture processes, it must be considered that capacity utilization can be increased by changing the DBC value. Indeed, higher DBC values can be obtained by steepening the breakthrough curve. This can be achieved by decreasing the loading ï¬ow velocity. However, lowering the loading flow velocity negatively impacts productivity, which will be unavoidably decreased (besides, buffer consumption increases).
This trade-off can be alleviated by employing multicolumn countercurrent processes (7). One of the most modern approaches for the capture step in semi-continuous mode is captureSMB. In its simplest version, two identical columns (packed with Protein A resin in the case of mAb purification) are connected through a series of valves. It is a quite complex process that can be briefly summarized in the steps represented in Figure 3. Interested readers are referred to references (13–15) for a
comprehensive description.

Figure 3: Schematic representation of twin-column capture SMB process. CIP stands for cleaning-in-place.
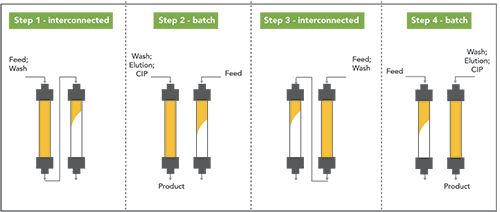
As it can be seen from Figure 3, there are moments when columns are sequentially loaded and washed (so-called interconnected steps), and others where columns are not connected to each other (batch steps). During batch steps, one column is washed, eluted, and regenerated, while loading is continued on the other. A full cycle is completed when the two columns turn back in their initial position. What is worth mentioning is that captureSMB makes it possible to drastically increase capacity utilization. A schematic representation is given in Figure 2, where a hypothetical breakthrough curve is represented. In batch chromatography, only the mass represented by area A is loaded on the column. This corresponds to the mass that can be loaded before 1% DBC.
In twin-column captureSMB, the loading can be increased. Therefore, the first column is loaded up to a X% DBC (usually 70% DBC), containing the mass corresponding to A + B in Figure 2, while mass in area C (breaking through from the first column) will be captured in the second column. The total A + B + D area corresponds to the maximum saturation capacity, Qsat. Thus, according to this scheme, capacity utilization for the two processes can be expressed, as reported in Table I.

As an example, captureSMB showed an increase of +26% in productivity and +11% in capacity utilization at a linear velocity of 150 cm/h for the purification of an IgG1 antibody on Amsphere JWT-203 protein A resin (16). The outcome was even better at 600 cm/h, with increases of +35% and +41% for productivity and capacity utilization, respectively. These results indicate a further advantage of captureSMB over batch processes, that is the possibility of operating at higher linear velocities since loadings are performed at much higher values than 1% DBC.
Another example is reported in (17), where mAb fragments have been purified in captureSMB by using a Capto L resin. Here, results showed a clear advantage of captureSMB over the correspondent batch process by achieving a +60% increase in loading, a +93% higher productivity, and a -54% in buffer consumption.
Multicolumn Countercurrent Solvent Gradient Purification (MCSGP)
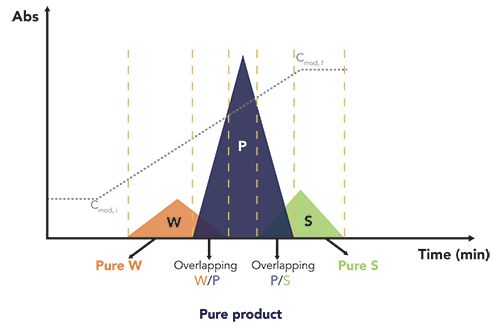
Differently from the capture step, polishing is needed to remove all product-related impurities, including, but not limited to, isoforms, truncates, aggregates, and deamidates. These impurities are usually produced during the synthesis, and they usually have very similar chromatographic characteristics to those of the target. The presence of product-related impurities can generate several peak overlapping regions in the chromatogram, where slightly weaker, W, and slightly stronger, S, adsorbing impurities are co-eluted with the front and the rear part of the peak of the target product, P (see Figure 4). In these cases, batch purifications are most likely governed by a yield-purity trade-off. This means that, in order to obtain a pool with acceptable purity for pharmaceutical standards, the collection window need to be narrowed at the cost of yield (and vice versa). To avoid wasting considerable amounts of target product, the overlapping regions (where the target component is still present but with an excessive amount of impurities) are manually recycled and reprocessed. This is a very labor-intensive activity that tremendously impacts on the productivity of the process.

Figure 4: Schematic illustration of a ternary separation where the chromatographic peak of the target product (P) partially overlaps with those of two product related impurities. Here W refers to weakly adsorbing impurities and S to strongly adsorbed ones. Dotted grey line represents a hypothetical gradient of the modifier from an initial concentration Cmod, I to a final concentration Cmod, f.
The yield-purity trade-off can be alleviated by employing multicolumn countercurrent techniques. Among these, the multicolumn countercurrent solvent gradient purification (MCSGP) is a semi-continuous process suitable for the challenging purification of complex mixtures, that also permits the use gradient elution (18–21). This is particularly interesting for the separation of large biomolecules, whose retention is strongly affected by the organic modifier concentration (20,22–24). In Figure 3, the principles of MCSGP, in the case of a ternary separations, are schematically depicted. As in captureSMB, also in MCSGP two (or more) identical columns are used.
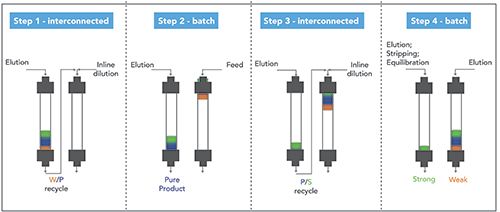
Differently from captureSMB, where recycling occurs during loading, in MCSGP instead recycling takes place during elution (see Figure 5). Indeed, the feed is loaded on the first column, the overlapping regions (W/P and P/S) are recycled on the second column while the purest fraction of product (P) is collected from the first one. Then the second column is fed with fresh feed, in order to keep the loading constant, and the elution starts now from the second column to the first one. One cycle ends when the two columns turn back in their initial position. The process runs in a cyclic way, and a steady state is reached where purity and recovery do not change cycle after cycle. This mechanism partially overcomes the yield-purity trade-off usually faced in batch separations. Indeed, the recycling of overlapping regions can increase the yield of the collected product while maintaining purity that is at least equivalent to that of a batch process (product purity strictly depends on the pooling criteria). The interested reader can find a detailed description of the process in references (12,25,26–28).
Figure 5: Schematic illustration of a twin-column MCSGP process.
MCSGP has been successfully applied to the purification of many classes of biomolecules. Different chromatographic media can be used in MCSGP, ranging from reversed-phase columns for the purification of peptides (25) to ion-exchange for the purification of oligonucleotides (29) or mAb charge variants (21,30).
It is worth mentioning that even a small increase in yield can be very advantageous when dealing with very expensive biopharmaceuticals. For example, references (21,30) report the purification of charged variants of mAbs with MCSGP on an ion-exchange column. An increase in yield of +56% and +74% was observed for the purification of bevacizumab (used for the treatment of many cancer diseases) and trastuzumab (used for the treatment of breast cancer), respectively, by maintaining purity constant with respect to batch purifications (30). Also, the purification process of oligonucleotides can be boosted through MCSGP. Indeed, the yield in the purification of a mixture of oligonucleotides on HiScreen Q Sepharose FFcolumns was increased from 60% to 91% by moving from batch to MCSGP, maintaining the purity at 92% (29). Recently, some of the authors of this paper have applied the MCSGP process to the purification of a therapeutic peptide from solid-phase synthesis on a C8 stationary phase (25), allowing for a +23% yield compared to the batch process, with an unchanged purity of 89%.
Conclusions and Future Perspectives
Continuous, or semi-continuous, countercurrent techniques make it possible to partially overcome common limitations of current single-column purification strategies that often represent a bottleneck of the whole production process. CaptureSMB makes it possible to increase both capacity utilization of the resin and productivity for the capture process, making it possible to operate also at faster linear velocities than correspondent batch processes. This technique is particularly suitable for the purification of mAbs on Protein A stationary phases, but it can be used with any other affinity system (for example, protein-ligand). On the other hand, MCSGP permits to alleviate the yield-purity trade-off typical of polishing batch processes by allowing for the internal recycling of overlapping regions of the chromatogram where the target is still present in a considerable amount but polluted with impurities. This technique has been successfully applied for the purification of peptides, oligonucleotides, and charge variants of mAbs, but it can be used for any other class of biomolecules.
The greatest advantage of these techniques is that, once the experimental conditions have been optimized, the purification process can be completely automated. Therefore, no human intervention is required to process large quantities of material.
Thanks to these advantages, multicolumn countercurrent techniques represent a convenient alternative over traditional batch purification processes for the ongoing development of novel therapeutics, vaccines, and monoclonal antibody therapies for the treatment of many diseases, including pandemic COVID-19.
Acknowledgments
The authors thank the Italian University and Scientific Research Ministry (grant PRIN2017Y2PAB8003, “Cutting edge analytical chemistry methodologies and bio-tools to boost precision medicine in hormone-related diseases”).
References
- G. Li and E. De Clercq, Nat. Rev.19, 149–150 (2020).
- J.S. Morse, T. Lalonde, S. Xu, and W.R. Liu, ChemBioChem 21, 730–738 (2020).
- C. Liu, Q. Zhou, Y. Li, L.V. Garner, S.P. Watkins, L.J. Carter, J. Smoot, A.C. Gregg, A.D. Daniels, S. Jervey, and D. Albaiu, ACS Cent. Sci. 6, 315–331 (2020).
- E. Nicastri, N. Petrosillo, T. Ascoli Bartoli, L. Lepore, A. Mondi, F. Palmieri, G. D’Offizi, L. Marchioni, S. Muratelli, G. Ippolito, and A. Antinori, Infect. Dis. Rep. 12:8543, 3–9 (2020).
- A.A. Shukla and J. Thömmes, Trends Biotechnol. 28, 253–261 (2010).
- G. Guiochon, A. Felinger, A. Katti, and D. Shirazi, Fundamentals of Preparative and Nonlinear Chromatography (Academic Press, Boston, Massachusetts, 2nd Ed., 2006).
- G. Subramaniam, Continuous Processing in Pharmaceutical Manufacturing (WileyâVCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 1st Ed., 2015).
- M. Mazzotti, G. Storti, and M. Morbidelli, J. Chromatogr. A 769, 3–24 (1997).
- E. R. Francotte and P. Richert, J. Chromatogr. A1997, 101–107 (1997).
- H. Schramm, M. Kaspereit, A. Kienle, and A. Seidel-Morgenstern, J. Chromatogr. A 1006, 77–86 (2003).
- [T. Kröber, M. W. Wolff, B. Hundt, A. Seidel-Morgenstern, and U. Reichl, J. Chromatogr. A 1307, 99–110 (2013)
- B. Kelley, Biotechnol. Progr. 23, 995–1008 (2007).
- F. Steinebach, T. Müller-Späth, and M. Morbidelli, Biotechnol. J. 11, 1126–1141 (2016).
- J. Angelo, J. Pagano, T. Müller-Späth, K. Mihlbachler, S. Chollangi, X. Xu, S. Ghose, and Z. J. Li, BioProcess Intl. 16(4), 1–6 (2015).
- M. Angarita, T. Müller-Späth, D. Baur, R. Lievrouw, G. Lissens, and M. Morbidelli, J. Chromatogr. A1389, 85–95 (2015).
- T. Müller-Späth, M. Angarita, D. Baur, M. Morbidelli, R. Lievrouw, M. Bavand, G. Lissens, and G. Strohlein, Biopharm Int. 26 (2013).
- C. Grossman, G. Ströhlein, M. Morari, and M. Morbidelli, J. Proc. Control20, 618–443 (2010).
- W. Jin and P. C. Wankat, Ind. Eng. Chem. Res.44, 1565–1575 (2005).
- A. L. Zydney, Biotech. Bioeng.113, 465–475 (2016).
- G. Ströhlein, L. Aumann, M. Mazzotti, and M. Morbidelli, J. Chromatogr. A1126, 338-346 (2006).
- T. Müller-Späth, L. Aumann, L. Melter, G. Ströhlein, and M. Morbidelli, Biotech. Bioeng.100, 1166–1177 (2008).
- N. Marchetti, F. Dondi, A. Felinger, R. Guerrini, S. Salvadori, and A. Cavazzini, J. Chromatogr. A1079, 162–172 (2005).
- D. Åsberg, M. Lésko, T. Leek, J. Samuelsson, K. Kaczmarski, and T. Fornstedt, Chromatographia80, 961–966 (2017).
- C. De Luca, S. Felletti, M. Macis, W. Cabri, G. Lievore, T. Chenet, L. Pasti, M. Morbidelli, A. Cavazzini, M. Catani, and A. Ricci, J. Chromatogr. Ahttps://doi.org/10.1016/j.chroma.2019.460789 (2020).
- C. De Luca, S. Felletti, G. Lievore, A. Buratti, S. Vogg, M. Morbidelli, A. Cavazzini, M. Catani, M. Macis, A. Ricci, and W. Cabri, submitted to J. Chromatogr. A (2020).
- D. Pï¬ster, L. Nicoud, and M. Morbidelli, Continuous Biopharmaceutical Processes-Chromatography: Bioconjugation and Protein Stability (Cambridge Series in Chemical Engineering, Cambridge University Press, Cambridge, UK, 2018).
- L. Aumann and M. Morbidelli, Biotech. Bioeng.98, 1043-1055 (2007).
- [28] F. Steinebach, N. Ulmer, L. Decker, L. Aumann, and M. Morbidelli, J. Chromatogr. A 1492, 19–26 (2017).
- Application Note. P2.V1, https://www.chromacon.com/resources/public/lava3/media/kcfinder/files/Oligonucleotide_MCSGP_application_note.pdf (2019).
- T. Müller-Späth, M. Krättli, L. Aumann, G. Ströhlein, and M. Morbidelli, Biotech. Bioeng. 107, 652–662 (2010).
Chiara De Luca, Simona Felletti, Giulio Lievore, Alessandro Buratti, Alberto Cavazzini and Martina Catani are with the Dept. of Chemistry and Pharmaceutical Sciences, at the University of Ferrara, in Ferrara, Italy. Mattia Sponchioni is with the Dept. of Chemistry, Materials and Chemical Engineering at the Politecnico di Milano, in Milan, Italy. Marco Macis, Antonio Ricci and Walter Cabri are with the Fresenius Kabi iPSUM, in Rovigo, Italy. Walter Cabri is also with the Department of Chemistry at the University of Bologna, in Bologna, Italy. Direct correspondence to: martina.catani@unife.it

![[1]L.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F2ca2ff2694c34a129d7ae348d24f6041f6ff9fdf-500x129.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "[1]L.jpg")


![[1]L1593120170948.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F77f8f859bb7a2582ccded7f5dd0b4cc0efde35c1-500x137.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "[1]L1593120170948.jpg")






, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

