Basics of HPLC Peptide Analysis
Fundamentals aspects of using HPLC techniques for protein analysis are described as they relate to the analysis of post-translational modifications (PTMs) and glycoprofiles.
Much information is available when biomolecules are analyzed at the protein level, such as molecular weight, structural integrity, charge variants, aggregation, and post-translational modifications (PTMs). However, identification of PTM modification sites, as well as other critical quality attributes such as the glycoprofile, requires digesting the protein into representative peptides using a suitable proteolytic digestion enzyme.
The digested peptide-containing solution is then chromatographed, commonly using a generic reversed-phase liquid chromatography (LC) methodology that consists of an acidic mobile phase, a steeper gradient over a wider range, and a longer alkyl chain stationary phase (such as C18, for example) as compared to the method employed to analyze an intact protein.
A typical peptide map of a digested monoclonal antibody (mAb) is shown in Figure 1. It is considerably more complex than those generated for intact proteins, due to the number of peptides liberated and the artifacts that arise from the digestion process, such as residual reagents and missed cleavages.


Figure 1: Typical reversed-phase chromatogram of a mAb peptide map.
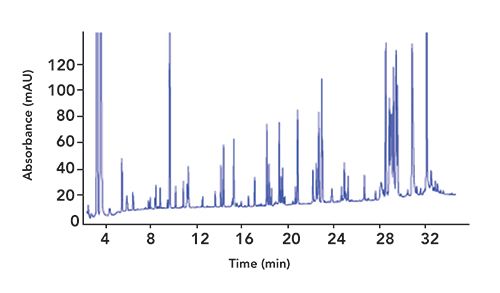
Great care and consideration are required during the digestion process, as the proteolytic enzymes used and the conditions employed (pH, temperature, even storage time) not only affect the overall number of peptides liberated, but also the stability of associated PTMs, and can even introduce protein modifications of their own.
Broadly speaking, the digestion process can be broken down into three separate steps: reduction, alkylation, and digestion.
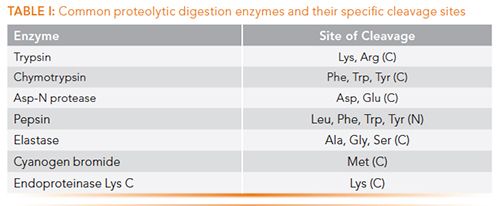
The first stage in the reduction step is to denature the mAb. This is commonly accomplished with an acid-labile surfactant that removes the higher order structure of the protein and exposes many otherwise internal disulfide bonds. These disulfide bonds are then ready for reduction, which is achieved using dithiothreitol. The pH is maintained at physiological levels throughout the process using buffers. To prevent reformation of disulfide bridges across the thiol groups of the cysteine (C) residues, the protein is then incubated with an alkylating agent such as 2-iodoacetamide, once again at physiological pH. The final stage is the addition of a proteolytic agent, which is capable of site-specific protein digestion. Table I details these enzymes and highlights their specific cleavage sites. Typically, fewer cleavage sites leads to larger, and therefore, fewer resulting peptides, and vice versa.


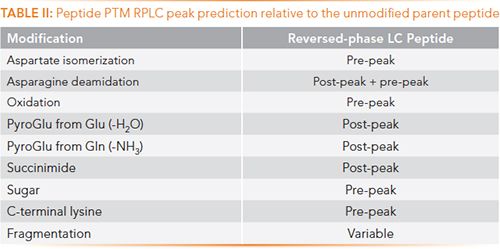
Due to the precise and predictable nature of the hydrophobic retention of reversed-phase LC, estimates as to where the modified peptide will elute in relation to the native, unmodified variant can be made (Table II). This can be a helpful tool when trying to identify and assign unexpected peaks. Asparagine deamidation can produce both pre- and post-peaks, due to deamidation occurring via the succinimide intermediate, iso-Asp (pre-peak) and Asp (post-peak) in a 3/4: 1 ratio.








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")


Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.

, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

