Analytical Method Validation: Back to Basics, Part I
LCGC North America
The authors present the first installment in their examination of the basics of method validation.
In any regulated environment, analytical method validation (AMV) is a critical part of the overall process of validation. AMV is a part of the validation process that establishes, through laboratory studies, that the performance characteristics of the method meet the requirements for the intended analytical application and provides an assurance of reliability during normal use; sometimes referred to as "the process of providing documented evidence that the method does what it is intended to do." Regulated laboratories must perform AMV to be in compliance with government or other regulators, in addition to being good science. A well-defined and documented validation process can provide evidence not only that the system and method is suitable for its intended use, but can aid in transferring the method and satisfy regulatory compliance requirements.


Michael Swartz
Validation is also the foundation of quality in the high performance liquid chromatography (HPLC) laboratory, and AMV is just one part of a regulatory quality system that incorporates both quality control and quality assurance (1,2). The terms "quality control" and "quality assurance" often are used interchangeably, but in a properly designed and managed quality system, the two terms have separate and distinct meanings and functions. Quality assurance (QA) can be thought of as related to process quality; whereas quality control (QC) is related to the quality of the product. In a given organization, it does not matter what the functions are named, but the responsibilities for these two activities should be defined clearly. Both quality assurance and quality control make up the Quality Unit, and are essential to the production of analytical results that are of high quality and are compliant with the appropriate regulations.


Ira Krull
QC is the process that determines the acceptability or unacceptability of a product or a product plan, and is determined by the comparison of a product against the original specifications that were created before the product existed. In some organizations, the QC group is responsible for the use of the method to perform analysis of a product. Other tasks related to QC can include documented reviews, calibrations, or additional types of measurable testing (such as sampling) and will reoccur more often than activities associated with quality assurance. QC usually will require the involvement of those directly associated with the research, design, or production of a product. For example, in a laboratory-notebook peer-review process a QC group would check or monitor the quality of the data, look for transcription errors, check calculations, verify notebook sign-offs, and so forth.
QA is determined by top-level policies, procedures, work instructions, and governmental regulations. At the beginning of the validation process, QA can provide guidance for the development of or review of validation protocols and other validation documents. During the analytical stage, QA's job is to ensure that the proper method or procedure is in use and that the quality of the work meets the guidelines and regulations. QA can be thought of as the process that will determine the template and pattern of quality control tasks. As opposed to QC checks, QA reports are more likely to be performed by managers, by corporate level administrators, or third-party auditors through the review of the quality system, reports, archiving, training, and qualification of the staff that performs the work.
AMV Guidance
Since the late 1980s, government and other agencies (for example, FDA, International Conference on Harmonization-ICH) have issued guidelines on validating methods. In 1987, the FDA designated the specifications in the current edition of the United States Pharmacopeia (USP) as those legally recognized when determining compliance with the Federal Food, Drug, and Cosmetic Act (3,4). More recently, new information has been published, updating the previous guidelines and providing more detail and harmonization with International Conference on Harmonization (ICH) guidelines (5,6). Guidelines are documents prepared for both regulatory agency personnel and the public that establish policies intended to achieve consistency in the agency's regulatory approach, and to establish inspection and enforcement policies and procedures. For example, the FDA guidance provides recommendations to applicants on submitting analytical procedures, validation data, and samples to support the documentation of the identity, strength, quality, purity, and potency of drug substances and drug products, and it is intended to assist applicants in assembling information, submitting samples, and presenting data to support analytical methodologies. The recommendations apply to drug substances and drug products covered in new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), product license applications (PLAs), and supplements to these applications.
One final introductory comment: Although for the most part the topic of discussion in most "Validation Viewpoint" columns is HPLC, the guidelines are generic; that is, they apply to any analytical procedure, technique, or technology used in a regulated laboratory (for example, gas chromatography [GC], mass spectrometry [MS], or IR spectroscopy). It also should be noted that the USP publishes official methods, often called compendial methods, which have been accepted by the USP as already validated. However it is common practice to verify these methods, and a separate USP chapter is devoted to this topic (7,8).
The Validation Process
When looking at the guidelines, one observes that AMV is just one part of the overall validation process that encompasses at least four distinct steps: software validation, hardware (instrumentation) validation–qualification, analytical method validation, and system suitability. The overall validation process begins with validated software and a validated–qualified system; then a method is developed and validated using the qualified system. Finally, the whole process is wrapped together using system suitability. Each step is critical to the overall success of the process.
Software Validation
A comprehensive treatment of software validation is outside the scope of this column. However, it is an important topic to at least touch on here as these days, every modern HPLC laboratory makes use of computerized systems to generate and maintain source data and documentation from a variety of instrumentation. These data must meet the same fundamental elements of data quality (for example, attributable, legible, contemporaneous, original, and accurate) that are expected of paper records and must comply with all applicable statutory and regulatory requirements. Two FDA guidelines have appeared recently that address the topic of software validation, and should be consulted for more detailed information (9,10). In addition, in March 1997, the FDA issued 21 CFR part 11, which provided the original criteria for acceptance of electronic records, electronic signatures, and handwritten signatures executed to electronic records as equivalent to paper records and handwritten signatures executed on paper under certain circumstances (11). However, after the effective date of 21 CFR part 11, significant concerns regarding the interpretation and implementation of part 11 were raised by both FDA and the pharmaceutical industry, and as a result, 21 CFR part 11 was re-examined (12). The new Scope and Application Guidance clarified that the FDA intends to interpret the scope of part 11 narrowly and to exercise enforcement discretion with regard to part 11 requirements for validation, audit trails, record retention, and record copying. However, most of the other original Part 11 provisions remain in effect.


Table I: A time line approach to AIQ
Analytical Instrument Qualification
Before undertaking the task of method validation, it is necessary to invest some time and energy up-front to ensure that the analytical system itself is validated, or qualified. Qualification is a subset of the validation process that verifies proper module and system performance before the instrument being placed on-line in a regulated environment. In March, 2003, the American Association of Pharmaceutical Chemists (AAPS), the International Pharmaceutical Federation (FIP), and the International Society for Pharmaceutical Engineering (ISPE) cosponsored a workshop entitled "A Scientific Approach to Analytical Instrument Validation" (13). Among other objectives, the various parties (the event drew a cross-section of attendees; users, quality assurance specialists, regulatory scientists, consultants, and vendors) agreed that processes are "validated" and instruments are "qualified," finally reserving the term validation for processes that include analytical methods–procedures and software development.
The proceedings of the AAPS et al. committee have now become the basis for a new general USP chapter, number 1058, on Analytical Instrument Qualification (AIQ) that originally appeared in the USP's Pharmacopeial Forum (14–16). The chapter details the AIQ process, data quality, roles and responsibilities, software validation, documentation, and instrument categories.
Instruments are qualified according to a stepwise process grouped into four phases: design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ). The DQ phase usually is performed at the vendor's site, where the instrument is developed, designed, and produced in a validated environment according to good laboratory practices (GLP), current good manufacturing practices (cGMP), and ISO 9000 standards.
During the IQ phase, all of the activities associated with properly installing the instrument (new, pre-owned, or existing) at the users' site are documented. After the IQ phase is completed, testing is done to verify that the instrument and instrument modules operate as intended in an OQ phase. First, fixed parameters, for example, length, weight, height, voltage inputs, pressures, and so forth are either verified or measured against vendor-supplied specifications. Because these parameters do not change over the lifetime of the instrument, they usually are measured just once. Next, secure data handling is verified. Finally, instrument function tests are undertaken to verify that the instrument (or instrument modules) meets vendor and user specifications.
Instrument function tests should measure important instrument parameters according to the instruments' intended use and environment. For LC, the following types of tests might be included: n pump flow rate
- detector wavelength accuracy
- gradient linearity
- injector precision, linearity, and accuracy
- detector linearity
- column oven temperature.
Relevant OQ tests should be repeated whenever the instrument undergoes major repairs or modifications.
After an IQ and an OQ have been performed, PQ testing is conducted. PQ testing should be performed under the actual running conditions across the anticipated working range. PQ testing should be repeated at regular intervals; the frequency depends upon such things as the ruggedness of the instrument, and the criticality and frequency of use. PQ testing at periodic intervals also can be used to compile an instrument performance history.


Figure 1
In practice, a known method with known, predetermined specifications is used to verify that all of the modules are performing together to achieve their intended purpose. In practice, OQ and PQ frequently blend together in a holistic approach, particularly for injector linearity and precision (repeatability) tests, which can be conducted more easily at the system level. For HPLC, the PQ test should use a method with a well-characterized analyte mixture, column, and mobile phase. Figure 1 shows an example of a "vendor" PQ test method that incorporates the essence of a holistic OQ and PQ test. Actual user PQ tests should incorporate the essence of the system suitability section of the general chromatography chapter 621 in the USP (15) to show suitability under conditions of actual use.
System Suitability
According to the USP, system suitability tests are an integral part of chromatographic methods (17). These tests are used to verify that the resolution and reproducibility of, for example, a chromatographic system, are adequate for the analysis to be performed. System suitability tests are based upon the concept that the equipment, electronics, analytical operations, and samples constitute an integral system that can be evaluated as a whole.
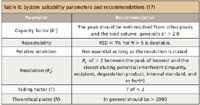
Table II: System suitability parameters and recommendations (17)
System suitability is the checking of a system to ensure system performance before or during the analysis of unknowns. Parameters such as plate count, tailing factors, resolution, and reproducibility (%RSD retention time and area for repetitive injections) are determined and compared against the specifications set for the method. However, samples consisting of only a single peak (for example, a drug substance assay in which only the API is present) can be used, provided that a column plate number specification is included in the method. Unless otherwise specified by the method, data from five replicate injections of the analyte are used to calculate the relative standard deviation if the method requires RSD > 2%; data from six replicate injections are used if the specification is RSD ≤ 2%. These parameters are measured during the analysis of a system suitability "sample" that is a mixture of main components and expected byproducts. Table II lists the terms to be measured and their recommended limits obtained from the analysis of the system suitability sample (18).
System suitability tests must be carried out before the analysis of any samples in a regulated environment. Following blank injections of mobile phase, water, or sample diluent, replicate system suitability injections are made, and the results compared against method specifications. If specifications are met, subsequent analyses can continue. If the method's system suitability requirements are not met, any problems with the system or method must be identified and remedied (perhaps as part of a formal out-of-specification (OOS) investigation), and passing system-suitability results must be obtained before sample analysis is resumed. To provide confidence that the method runs properly, it is also recommended that additional system suitability samples (quality control samples or check standards) are run at regular intervals (interspersed throughout the sample batch); %-difference specifications should be included for these interspersed samples, to make sure the system still performs adequately over the course of the entire sample run. Alternatively, a second set of system-suitability samples can be included at the end of the run. Quality control check samples are run to make sure the instrument has been calibrated properly or standardized. Instrument calibration ensures that the instrument response correlates with the response of the standard or reference material. Quality control check samples also are used often to provide an in-process assurance of the test's performance during use. In general, AIQ and analytical method validation generally ensure the quality of analysis before conducting a test; system suitability and quality control checks ensure the quality of analytical results immediately before or during sample analysis.


Table III: Maximum specifications for adjustments to HPLC operating conditions
As previously mentioned, USP compendial methods are considered to be validated, however, adjustments to USP methods are allowed to meet system-suitability requirements; such instructions can be included in individual monographs. But method changes usually require some degree of revalidation. So an important question is, "At what point does an adjustment become a change or modification" to the method? Historically, if adjustments to the method are made within the boundaries of any robustness studies that were performed, no further actions are warranted as long as system-suitability criteria are satisfied. Unfortunately, the robustness information is not readily available for many USP methods, and any adjustment outside of the bounds of the robustness study constitutes a change to the method and requires a revalidation.
In 1998, Furman and colleagues proposed a way to classify allowable adjustments (19). But it was not until 2005 that guidance appeared on the topic (20–22). Although USP guidance on this topic recently was included into USP Chapter 621 on chromatography (17), the FDA Office of Regulatory Affairs (ORA) has had guidance in place for a number of years (20). Table III summarizes the adjustments allowed for various HPLC parameters taken from both the USP and ORA documents. Adjustments outside of the ranges listed in Table III constitute modifications, or changes, which are subject to additional validation. Sound scientific reasoning should be used when determining method adjustment versus method change for a specific method. For example, if robustness studies have shown that the method conditions allow less variability for a parameter than that listed in Table III, or when robustness testing have shown that more variability is allowed, the robustness results (as summarized in the validation report) should prevail.
A few additional comments: Adjustments to chromatographic systems to comply with system-suitability requirements should not be made to compensate for system malfunctions or column failures. To prevent specification "creep," adjustments are made only from the original method parameters each time the method is run and are not subject to continuous adjustment. Adjustments are permitted only when suitable reference standards are available for all compounds used in the suitability test and only when those standards are used to show that the adjustments have improved the quality of the chromatography so as to meet system suitability requirements. The suitability of the method under the new conditions must be verified by assessment of the relevant analytical performance characteristics. Because multiple adjustments can have a cumulative effect in the performance of the system, any adjustments should be considered carefully before implementation.
Conclusion
In today's global market, validation can be a long and costly process, involving regulatory, governmental, and sanctioning bodies from around the world. A well-defined and documented validation process provides regulatory agencies with evidence that the system (instrument, software, method, and controls) is suitable for its intended use. AMV is a critical part of this process, and will be further addressed in detail in Part II of this two-part series.
Michael Swartz "Validation Viewpoint" Co-Editor Michael E. Swartz is Research Director at Synomics Pharmaceutical Services, Wareham, Massachusetts, and a member of LCGC's editorial advisory board.
Ira S. Krull "Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.
References
(1) United States Food and Drug Administration, Guideline for submitting samples and analytical data for methods validation, February 1997. US Government Printing Office: 1990-281-794:20818, or at www.fda.gov/cder/analyticalmeth.htm
(2) Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding Of Drugs, 21 CFR Part 210, http://www.fda.gov/cder/dmpq/cgmpregs.htm
(3) Current Good Manufacturing Practice for Finished Pharmaceuticals, 21 CFR Part 211, http://www.fda.gov/cder/dmpq/cgmpregs.htm
(4) USP 32-NF 27, August 2009, Chapter 1225.
(5) Analytical Procedures and Method Validation. Fed. Reg. 65(169), 52,776-52,777, 30 August 2000. See also: www.fda.gov/cder/guidance
(6) International Conference on Harmonization, Harmonized Tripartite Guideline, Validation of Analytical Procedures, Text and Methodology, Q2(R1), November 2005, See www.ICH.org.
(7) USP 32-NF 27, August 2009, Chapter 1226.
(8) M.E. Swartz and I.S. Krull, LCGC 23(10), 1100–1109 (2005).
(9) FDA, General Principles of Software Validation; Guidance for Industry and FDA Staff. See: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm085371.pdf
(10) Guidance for Industry Computerized Systems Used in Clinical Investigations, FDA May 2007. See: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070266.pdf
(11) FDA, 21 CFR Part 11, "Electronic Records; Electronic Signatures; Final Rule." Federal Register Vol. 62, No. 54, 13429, March 20, 1997.
(12) FDA, Part 11, Electronic Records; Electronic Signatures — Scope and Application, 2003. See: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072322.pdf
(13) Qualification of Analytical Instruments for Use in the Pharmaceutical Industry: A Scientific Approach, AAPS PharmSciTech, 2004, 5(1) Article 22 (http://www.aapspharmscitech.org
(14) Pharmacopeial Forum , 31(5), 1453–1463 (Sept-Oct 2005).
(15) USP 32-NF 27, August 2009, Chapter 1058
(16) M.E. Swartz, Analytical Instrument Qualification, Pharmaceutical Regulatory Guidance Book (Advanstar Communications, July 2006), pp. 12–16.
(17) USP 32-NF 27, August 2009, Chapter 621
(18) Center for Drug Evaluation and Research (CDER), Reviewer Guidance: Validation of Chromatographic Methods, US Government Printing Office, 1994 - 615-023 - 1302/02757.
(19) W.B. Furman, J.G. Dorsey, and L.R. Snyder, Pharm. Technol. 22(6), 58–64 (1998).
(20) FDA ORA Laboratory Procedure, #ORA-LAB.5.4.5, USFDA ( 09/09/2005). See also: http://www.fda.gov/ora/science_ref/lm/vol2/section/5_04_05.pdf
(21) Pharmacopeial Forum, 31(3), 825 (May-June 2005).
(22) Pharmacopeial Forum, 31(6) 1681 (Nov.-Dec. 2005).
















University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.





