Analysis of Near 400 Pesticides in Tea via LC–MS/MS: Simple Sample Preparation and APCI to Improve Analyte Coverage


As regulatory laboratories search for and implement consolidated methods for multiple matrix and analyte classes, compound lists increase to hundreds or thousands of targets. Multi-instrument approaches are often relied upon to analyze all pesticide targets, with the workload split between liquid chromatography–mass spectrometry (LC–MS) and gas chromatography–mass spectrometry (GC–MS) instrumentation. In this work, a simple solvent extraction approach was coupled with dual source electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) modes on an LC–tandem mass spectrometry (MS/MS) instrument to analyze 395 analytes extracted from black tea (345 compounds via ESI and 50 compounds via APCI, along with the internal standards). Excellent method performance (defined as an accuracy of 70–120% and a precision of <20% at one of two validation levels) was achieved for over 93% of analytes, including compounds normally analyzed by GC–MS, such as trifluralin, chlorfenson, chlormephos, fenchlorphos, etridiazole, and others. This approach can allow the consolidation of a pesticide method to a single instrument or significantly reduce the workload of a complementary GC–MS method.
Because tea is the most consumed beverage worldwide, the global tea crop has immense cultural and economic importance (1). As with any crop, growers use pesticides to ensure high harvest yields and quality, resulting in consumer exposure via the brewing process (most notably for polar analytes such as neonicotinoids and organophosphates) (2–4). Regulatory and testing laboratories face challenges as consumer preference in organic products increases, pesticide screening lists broaden, and global tea trade and consumption grow. These laboratories rely on mass spectrometry (MS) techniques, such as gas chromatography–tandem MS (GC–MS/MS) and liquid chromatography–tandem MS (LC–MS/MS), to quantify pesticide targets, often requiring both instrument platforms to fully cover their target lists (5). As demonstrated in past publications, atmospheric pressure chemical ionization (APCI) in LC–MS/MS can substitute for GC–MS/MS in analyzing some nonpolar compounds that can reduce cost and complexity for routine analysis laboratories (6–8).
In the same efficiency vein, sample preparation techniques for tea analysis are often based around the quick, easy, cheap, effective, robust, and safe (QuEChERS) extraction techniques, solid-phase extraction (SPE), or both (9). These sample preparation approaches add complexity, cost, and time for minimal to no improvement in extraction efficiency, and matrix effects as compound panels are scaled up to hundreds or thousands of targets (10,11).
In the following work, a simple solvent extraction approach was coupled with a LC–MS/MS instrument fitted with a dual electrospray ionization (ESI) and APCI source to analyze approximately 400 pesticides extracted from black tea (345 compounds analyzed via ESI and 50 compounds determined via APCI). Excellent method performance (defined as accuracy of 70–120% and a precision of <20%) was achieved for over 93% of analytes, including compounds normally analyzed by GC–MS/MS, such as fluralins, chlormephos, chlorfenson, thiometon, and others (5).
Experimental
Materials
All experiments were carried out using a QSight LX50 ultrahigh-pressure LC (UHPLC) instrument coupled to a QSight 420 triple quadrupole MS instrument (PerkinElmer, Inc.) via a divert valve. All instrument control, method development, and data processing were completed using the Simplicity 3Q software. All pesticide standards were sourced in custom mixes from AccuStandard. The internal standards were sourced from the One Pesticide420 Reagent Kit (PerkinElmer, Inc.), containing 29 deuterated pesticide analogues. For sample extraction, 15 mL centrifuge tubes, a high- capacity vortex mixer (DVX-2500 from VWR International LLC), and a refrigerated centrifuge (5702 R from Eppendorf Canada Ltd) were used. Black tea, chamomile tea, herbal blend tea, and the catnip matrix were purchased from a local organic specialty store in Toronto, Ontario. The tea was ground prior to extraction by using a consumer-grade personal blender and sieved to a particle size of <1 mm. All the solvents and mobile phase additives that were used were LC–MS grade.
Analytical Protocols
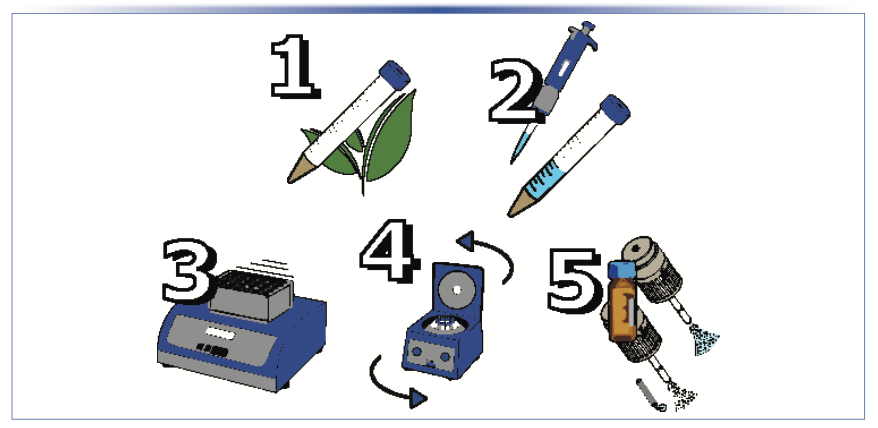
A solvent extraction sample preparation procedure (illustrated in Figure 1) with internal standard dilution was employed: 1 ± 0.01 g of the ground and sieved tea sample was weighed into a 15 mL centrifuge tube. For prespiked recovery experiments, pesticide standards were spiked into the solid matrix with the samples placed on a high-capacity vortex shaker pulsing at 1500 rpm for 1 hr. Following agitation, 10 μL of the internal standard mix was added, along with 5 mL of acetonitrile and 0.1% formic acid. After extracting the solvent addition, the samples were agitated again for 30 min at 1500 rpm. Following extraction, the samples were centrifuged for 5 min at >3 × 103 RCF, with 1 mL of the extract filtered through a 0.2 μm filter into a 2 mL amber vial for LC–MS/MS analysis.

 1 g of tea sample measured into a 15 mL centrifuge tube along with internal standards; 2) 5 mL of acetonitrile with 0.1% formic acid added; 3) sample agitated for 30 min during extraction; and 4) sample centrifuged before 5) filtration and analysis via LC–MS/MS with ESI and APCI methods.")
FIGURE 1: Analytical procedure employed for analyzing pesticides from tea: 1) 1 g of tea sample measured into a 15 mL centrifuge tube along with internal standards; 2) 5 mL of acetonitrile with 0.1% formic acid added; 3) sample agitated for 30 min during extraction; and 4) sample centrifuged before 5) filtration and analysis via LC–MS/MS with ESI and APCI methods.
Solvent calibrants with stable isotope-labeled internal standards were used for calibration, with nine calibration standards prepared in a neat solvent spanning the range of 0.1–250 ng/mL. The accuracy and precision of the method were determined at the 10 ng/g and 100 ng/g level (spiked in tea samples) across four sample replicates. Limits of quantification (LOQs) were reported at the lowest concentration level, which met the accuracy qualifiers within 70–120%, a relative standard deviation (RSD) of <20%, and the signal-to-noise (S/N) ratio of greater than 10 (for the quantifier transition) requirements. For analytes with accuracy values outside the range of 70–120%, LOQs were determined as the lowest point in the calibration curve with a S/N of greater than 10. Matrix effects were determined by comparing the slopes between calibration curves made in neat solvent and tea extract (with stock standards spiked into solvent and extract), which was reported as a percentage of signal suppression and enhancement (%SSE). Absolute matrix effects in the additional tea matrices of chamomile, herbal blend, and catnip were determined by comparing the peak areas between the post-extract spike and solvent-only standards at 10 ng/mL and 100 ng/mL, expressed as %SSE (12).
Instrumental Conditions
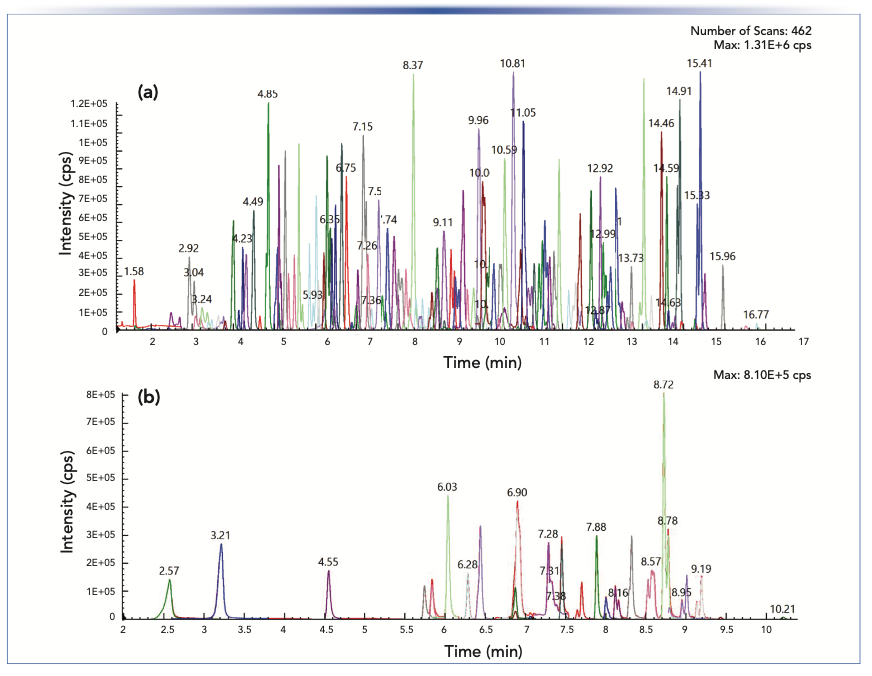
Instrumental parameters for both the APCI and ESI methods are shown in Table I. A time-managed multiple reaction monitoring (TM-MRM) method was built for both APCI and ESI compounds. In total, 690 MRM transitions were monitored for 345 compounds in the ESI method, and 100 MRM transitions were monitored for 50 compounds in the APCI method, along with the internal standards (shown in Figure 2). For both LC methods, a 100 mm x 4.6 mm i.d. 2.7 μm Quasar SPP C18 column (PerkinElmer, Inc.) was used.



 ESI and (b) APCI methods, respectively.")
FIGURE 2: TIC of 345 and 50 pesticides analyzed using (a) ESI and (b) APCI methods, respectively.
Results and Discussion
LC–MS/MS Method Performance
Because of the high sample processing load of routine testing facilities, several method development decisions were made to meet their high sample throughput requirements. The pursuit of a protocol-based around solvent extraction, a commercially available internal standard mixture, and internal standard calibration resulted in an easy-to-execute analysis procedure. Negligible matrix effects (80 < SSE < 120%) were observed across 94% (371 compounds) of the compound panel. The remaining 6% of compounds were either suspected to be degrading in the extract (thiocyclam and diafenthiuron) (13,14), or displayed signal suppression or enhancement greater than 20% (such as dodemorph and chlormephos, respectively). These compounds with matrix effects greater than 20% demonstrated the need to incorporate internal standards into the calibration and sample preparation procedure to improve method accuracy, as well as to provide flexibility to expand the protocol to additional matrices and compounds in the future. Internal standards were added before extraction to correct for both losses in the sample preparation procedure along with matrix effects during the analysis step. Excellent analytical figures of merit were observed for most of the analytes under study (displayed for all compounds of interest in Table II), with %SSE in black tea ranging from 161 to 69%. Black tea has been referenced as a difficult matrix in regard to suppression by co-extracting matrix-sourced interferences via QuEChERS and other dilute-and-shoot methods. However, extensive suppression was not observed in our implementation (15–17).


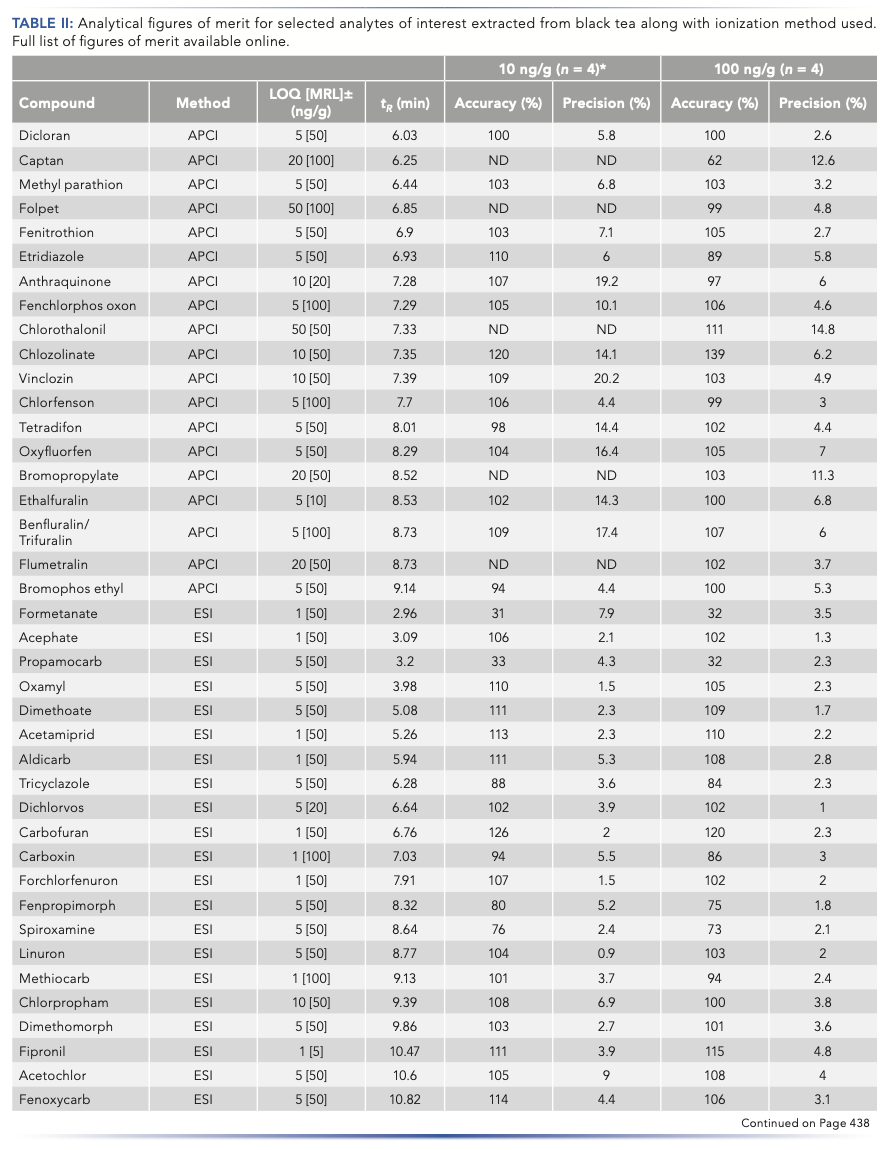


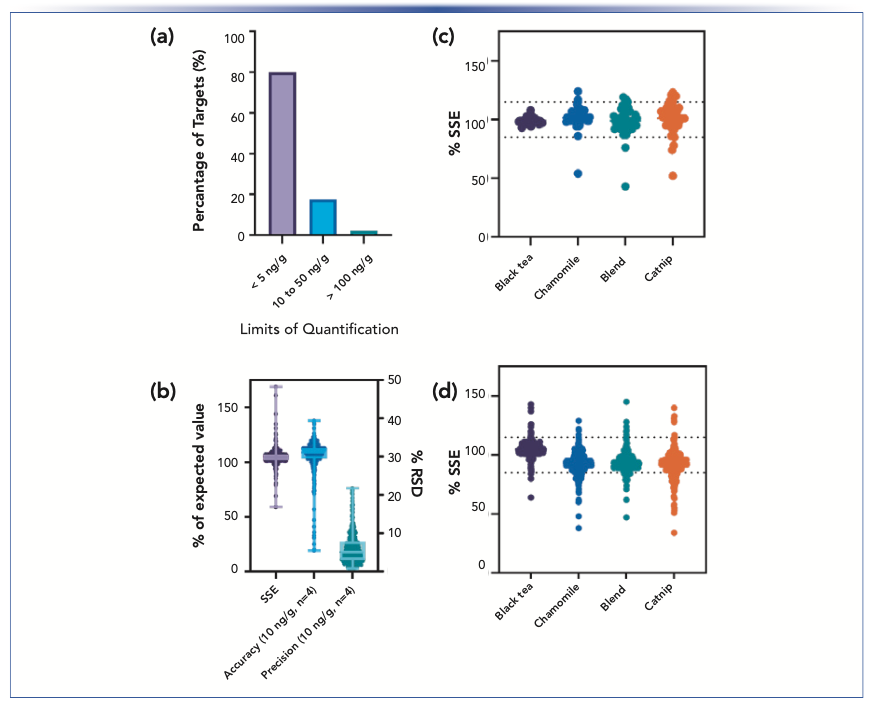
Similarly, accuracy, precision, and LOQs were determined for the compound panel across both LC methods. Of the 395 compounds validated, 93% (373 compounds) displayed accuracy values of 70–120% along with sub-20% RSD values at the 100 ng/g validation level with 3% (15 compounds) within 60–140%. Full compound panel validation results, retention times (tR), and method details are presented in Table II with a summary of the Table II results displayed in Figure 4 for %SSE, accuracy, and precision data at the 10 ng/g validation level. Of the 11 compounds that resulted in recoveries below 60%, eight of them (dodine, formetanate, propamocarb, ethirimol, imazalil, dodemorph, pymetrozine, and cyromazine) were found to have constant recoveries at five concentration levels spanning 5–500 ng/g (cyromazine at 100–500 ng/g), with reproducibility across both experiments. Recoveries outside of the target range of 70–120% can be accounted for with a correction factor and if instrumental sensitivity remains sufficient (LOQs for the aforementioned compounds are <10 ng/g with recovery losses). The two remaining analytes of diafenthiuron and thiocyclam with a recovery lower than 20% are known to be pro-insecticides, with degradation integral to their mode-of-action. Therefore, if surveillance of these compounds is critical, it is recommended to additionally monitor their degradation products: diafenthiuron, derived-carbodiimide, and nereistoxin, respectively (13,14,18).

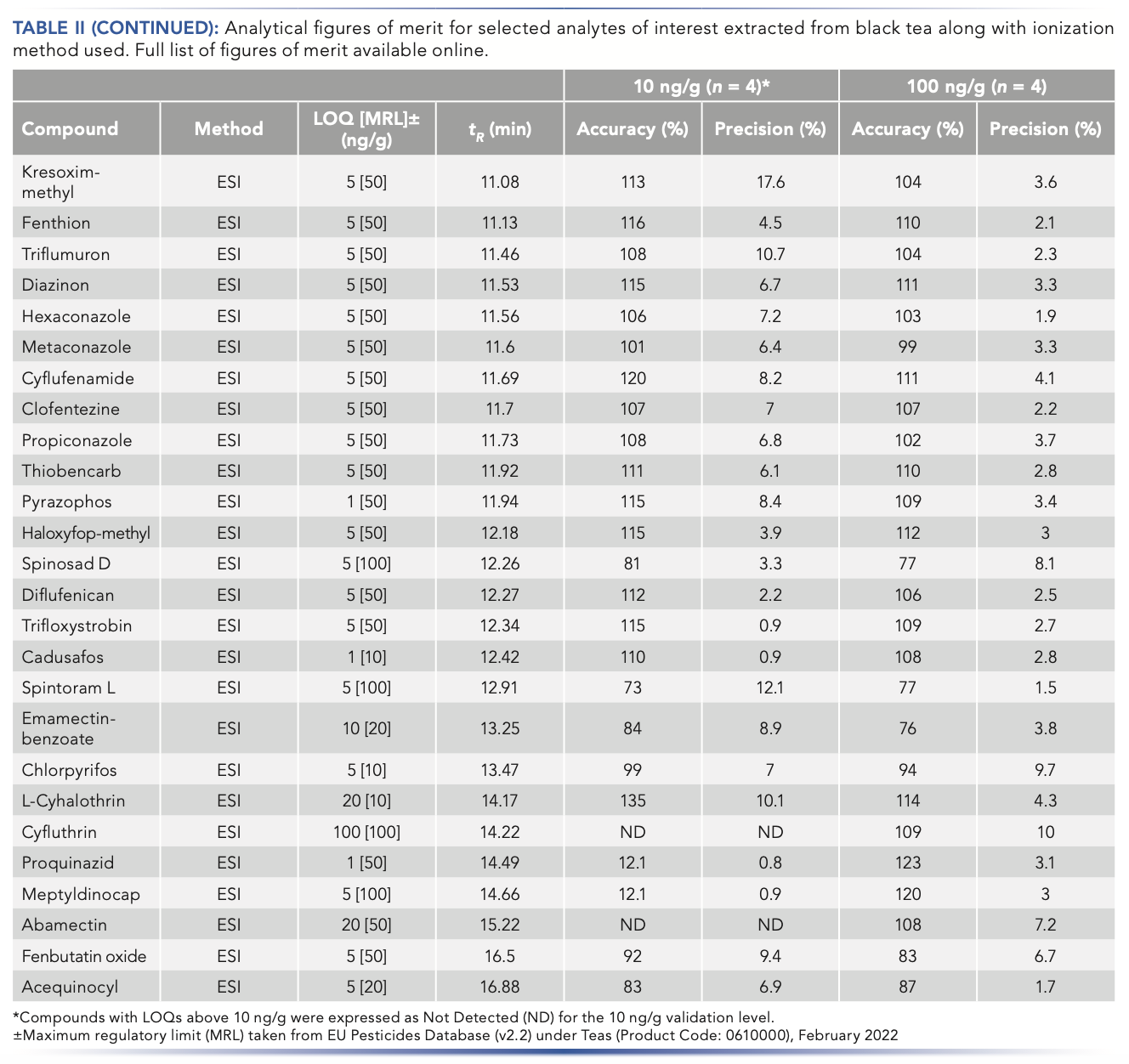
 chlormephos and (b) benfluralin. The extracts from the tea samples are compared with differing concentration levels and blank matrix injections.")
FIGURE 4: Comparisons of APCI-LC–MS/MS chromatograms for (a) chlormephos and (b) benfluralin. The extracts from the tea samples are compared with differing concentration levels and blank matrix injections.
Further Matrix Effects Investigation
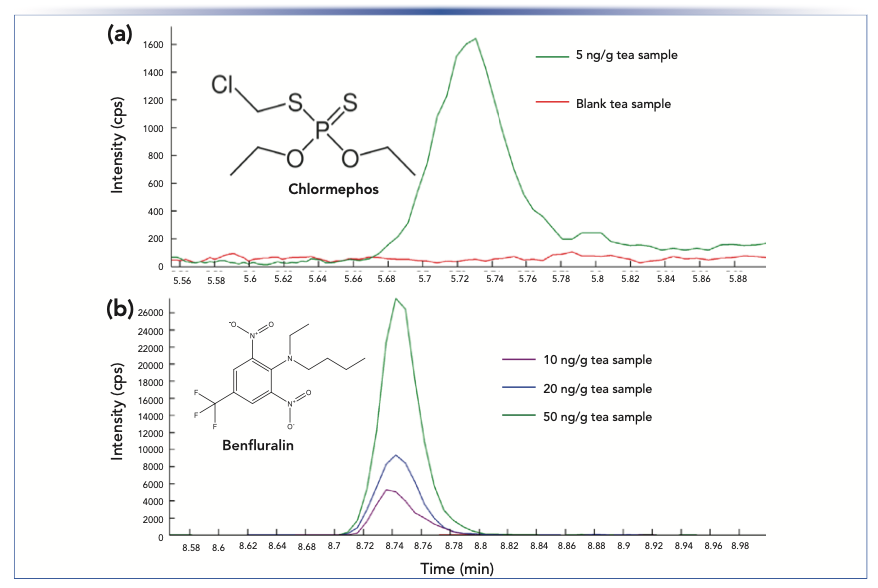
Following the promising method validation results in black tea, an extension of the matrix effects investigation was pursued to investigate the suitability of this approach in other herbal teas. An absolute matrix effects study was designed to explore method performance between black tea, chamomile, an herbal blend (containing chamomile, spearmint, lemongrass, blackberry leaves, orange blossoms, hawthorn, and rosebuds), and catnip (12). Samples were extracted following the procedure highlighted in Figure 1, and the extracts were spiked at 10 and 100 ng/mL with the pesticides under study. Peak areas were compared between the post-extract spikes and solvent-only samples, expressed as %SSE. The summarized results can be found in a scatter plot in Figure 3 for all compounds and matrices. The robustness of the separation method to matrix effects continued in the additional tea matrices tested. For the ESI method, 97% of analytes were found to have negligible matrix effects (defined as 85% < %SSE < 115%) in black tea, 93% in chamomile, 95% in the herbal blend, and 85% in catnip (12). The APCI method performed similarly with 100% of compounds in black tea displaying negligible matrix effects, 94% in chamomile and the herbal blend, and 89% in catnip. The low matrix effects across multiple herbal tea products suggests that the method developed can be adapted to analyze several types of tea with a trivial revalidation.

 Summary of select method validation data, with limits of quantification (LOQs) under 5 ng/g for approximately 80% of compounds under study, and (b) individual compound data points and corresponding box and whisker plots for matrix effects (%SSE), accuracy, and precision at the 10 ng/g validation point (for the compounds of which LOQ <10 ng/g). (c) Absolute matrix effects are summarized for APCI at 100 ng/mL for 50 compounds, and (d) for ESI at 10 ng/g for 342 compounds.")
FIGURE 3: (a) Summary of select method validation data, with limits of quantification (LOQs) under 5 ng/g for approximately 80% of compounds under study, and (b) individual compound data points and corresponding box and whisker plots for matrix effects (%SSE), accuracy, and precision at the 10 ng/g validation point (for the compounds of which LOQ <10 ng/g). (c) Absolute matrix effects are summarized for APCI at 100 ng/mL for 50 compounds, and (d) for ESI at 10 ng/g for 342 compounds.
LOQs Compared with Pesticide Maximum Regulatory Limits (MRLs)
For a method developed to be fit-for-purpose in the regulatory environment, it needs to meet regulatory performance levels. A complete comparison of all LOQs determined to MRLs was not possible, because not all compounds were regulated for use on tea consistently between regions. The most comprehensive and strict regulatory list of pesticides in tea was found via the European Commission. Of the 395 compounds validated, 227 compounds have been assigned an EU-harmonized MRL and are authorized for use in tea products. Upon comparison of the method LOQs, it was found that 172 of the 227 compounds had MRLs at least 10-fold greater than method LOQs (over 80% of analytes). The LOQs of the remaining 55 compounds were found to be less or equal to the MRL for all but azadirachtin and cyhalothrin.
Using APCI to Reduce ESI Workload and Expand Method Coverage
APCI has been shown to be less susceptible to matrix effects than ESI for bioanalytical and agrochemical analysis (19). Therefore, the addition of APCI to the protocol provides two primary benefits: it has the ability to monitor previously GC–MS/MS-only compounds (such as pentachloronitrobenzene, chlordane, fenitrothion, chlorfenapyr, and anthraquinone) as well as the flexibility to monitor compounds via APCI because of improved sensitivity compared to ESI (such as phthalimide-class compounds, parathion, fluralin-class compounds, and fenchlorphos), which is because of a different ionization mechanism or a reduction in matrix effects. In this work, samples were analyzed for the bulk of the analyte target list during the ESI method followed by a repeated sequence (injection) for APCI analysis of the 50 analytes that were not suitable for ESI. Low LOQs of 5 ng/g were achieved using APCI for compounds traditionally analyzed using GC–MS, such as fluralin-class compounds (benfluralin and profluralin), fenitrothion, chlorfenson, fenchlorphos oxon, dicloran, bromophos ethyl, and dichlofenthion. It is worth mentioning that the dual source setting for QSight LC–MS/MS allows for automatic LC method and MS probe switching without any manual intervention, which greatly reduces operation time and cost. Leveraging APCI has vastly simplified pesticide analysis in certain applications, and the work completed aims to broaden prior success (6–8).
Conclusion
With routine laboratories tasked with analyzing larger target lists and with greater numbers of samples requiring analysis, we deployed simple sample preparation protocols, such as solvent extraction, to meet the need of high sample throughput with lower cost. In this work, we demonstrated how a practical simple solvent extraction of black tea can be employed to analyze a large multi-residue panel of pesticides, with low limits of detection (with 319 of 395 analytes displaying LOQs of 5 ng/g or below), minimal matrix effects (94% of compounds with negligible %SSE in black tea), and excellent method performance (93% of compounds with accuracies within 70–120% and RSD <20%). Prior successes in cannabis matrices, along with this study, have suggested simple, acetonitrile extraction conditions and high-flow rate LC parameters allow for practical workflows when conducting pesticide analysis. Additionally, APCI analysis allowed for traditionally GC-only compounds (such as pentachloronitrobenzene, chlormephos, and others) to be included on this LC pesticide panel, reducing support instrument workload and potentially eliminating the need for a complementary GC–MS protocol for multiresidue pesticide quantification. We hope to extend these concepts to a plethora of food matrices of varying difficulties and compositions.
References
(1) S. Li, C.Y. Lo, M.H. Pan, C.S. Lai, and C.T. Ho, Food Funct. 4(1), 10–18 (2013).
(2) J. Wang, W. Cheung, and D. Leung, J. Agric. Food Chem. 62(4), 966–983 (2014).
(3) N. Manikandan, S. Seenivasan, M.N.K. Ganapathy, N.N. Muraleedharan, and R. Selvasundaram, Food Chem. 113(2), 522–525 (2009).
(4) M. Gupta and A. Shanker, Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 26(2), 157–163 (2009).
(5) L. Alder, K. Greulich, G. Kempe, and B. Vieth, Mass Spectrom. Rev. 25(6), 838–865 (2006).
(6) A. Dalmia, et al, Curr. Trends Mass Spectrom. 18(3), 22–29 (2020).
(7) A. Dalmia, et al, Cannabis Sci. Technol. 1(3), 38–50 (2018).
(8) A. Dalmia, et al, Appl. Note, 9 (2019). at <https://resources.perkinelmer.com/lab-solutions/resources/docs/APP_Canadian-Cannabis-Pesticide.pdf>
(9) G. Chen, P. Cao, and R. Liu, Food Chem. 125(4), 1406–1411 (2011).
(10) D. Steiner, M. Sulyok, A. Malachová, A. Mueller, and R. Krska, J. Chromatogr. A 1629, 461502 (2020).
(11) H.G.J. Mol, et al, Anal. Chem. 80(24), 9450–9459 (2008).
(12) B.K. Matuszewski, M.L. Constanzer, and C.M. Chavez-Eng, Anal. Chem. 75(13), 3019–3030 (2003).
(13) Y.S. Keum, J.H. Kim, Y.W. Kim, K. Kim, and Q.X. Li, Pest Manag. Sci. 58(5), 496–502 (2002).
(14) R. López-Ruiz, et al, Food Chem. 344, 128729 (2021).
(15) A. Malachová, M. Sulyok, E. Beltrán, F. Berthiller, and R. Krska, J. Chromatogr. A 1362, 145–156 (2014).
(16) H. Stahnke, S. Kittlaus, G. Kempe, and L. Alder, Anal. Chem. 84(3), 1474–1482 (2012).
(17) B. Greer, O. Chevallier, B. Quinn, L.M. Botana, and C.T. Elliott, TrAC - Trends Anal. Chem. 141, 116284 (2021).
(18) J.M. Clark, S.H. Lee, H.J. Kim, K.S. Yoon, and A. Zhang, Pest Manag. Sci. 57(10), 975–980 (2001).
(19) W.M.A. Niessen, P. Manini, and R. Andreoli, Mass Spectrom. Rev. 25(6), 881–899 (2006).
ABOUT THE AUTHORS
Alexander Kasperkiewicz and Feng Qin are with PerkinElmer, in Woodbridge, Ontario, Canada. Avinash Dalmia and Thomas Dillion are with PerkinElmer, in Shelton, Connecticut. Direct correspondence to: alexander.kasperkiewicz@perkinelmer.com


















Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.



Advances in Non-Targeted Analysis for PFAS in Environmental Matrices
March 27th 2025David Megson from Manchester Metropolitan University in Manchester, UK, spoke to LCGC International about the latest developments in non-targeted analysis (NTA) of per- and polyfluoroalkyl substances (PFAS) in environmental matrices based on a recent systematic review paper he has collaboratively published (1).

Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

