Chemistry in a Bottle: Ester Formation in Acidified Mobile-Phase Solvents


Acidified solvents are commonly used in reversed-phase liquid chromatography (RPLC) to adjust mobile-phase pH and the ionization state of ionogenic compounds. When methanol-containing solvents are acidified with formic or trifluoroacetic acids, esters can be formed at chromatographically relevant rates, affecting baseline quality, retention, and selectivity. Understanding these effects is the first step toward avoiding the problems that can be caused by esterification in the bottle.
In reversed-phase liquid chromatography (RPLC), mobile-phase additives are used for pH control because mobile-phase pH can strongly affect retention, peak shape, and method robustness for ionizable compounds. For instance, an amine-containing compound at a pH value lower than two units below the pKa of its protonated, acidic form (for example, the ammonium ion is the protonated acidic form of ammonia) is almost fully protonated (<0.1% present as neutral, unionized base). On the other hand, when the mobile-phase pH is more than two units higher than its pKa, the amine is nearly fully unprotonated and neutral (<0.1% present as protonated, positively charged acidic form), and when the pH is between these benchmarks (that is, [pKa − 2] < pH < [pKa + 2]), the ratio of the concentrations of the neutral and positively charged forms will be between 0.01 and 0.99. In RPLC, the protonated, positively charged form of the amine is less retained than the deprotonated, neutral form. Thus, the mobile-phase pH provides a powerful lever to adjust selectivity, with the most significant changes expected to occur within ±1 pH unit of the pKa of the compound (1). Nevertheless, peak shape (for example, peak tailing) for amine-containing compounds is strongly influenced by electrostatic interactions between the positively charged analytes and charged groups in the stationary phase, including the ionized silanol groups on the silica backbone of the reversed-phase (RP) columns (2). Thus, it is common practice to use a low mobile-phase pH for analyzing these amines so that the silica silanols are mostly protonated and neutral (and amines are fully protonated), and small changes in mobile-phase pH will not cause large changes in retention (3).
The type of organic solvent used in the mobile phase for RP separations can also have a significant effect on selectivity. Though acetonitrile is still the most commonly used solvent (4), methanol is often considered as a first alternative because it can yield significant differences in selectivity for some analyte-stationary phase combinations (5). Mass spectrometry (MS) detection continually gains in importance, and favors the use of volatile, low ionic strength additives to adjust the mobile-phase pH, such as formic acid (FA) or trifluoroacetic acid (TFA). FA is often preferred because it has been found that suppression of analyte ionization during the electrospray process is generally lower with FA compared to TFA (6). On the other hand, TFA has potential advantages over FA in some situations, including better peak shapes for amine-containing compounds, retention enhancement for highly hydrophilic amines because of ion-pairing, and higher acidity that yields lower mobile-phase pH at modest concentrations. However, carboxylic acids, such as FA, can react with alcohols like methanol to form esters, which causes a reduction in the acid content of the mobile phase. The kinetics of the esterification reaction in the specific case of FA and methanol were briefly studied by Przybytek and coworkers. They also demonstrated the potential for the process to affect the separation of peptides through changes in retention and resolution (7). Shifts in retention time can be problematic in two-dimensional LC (2D-LC), particularly when using heart-cutting 2D-LC mode. In this case, fractions of the first dimension (1D) effluent are often collected in reference to a previously collected 1D chromatogram. Any change in retention time because of the esterification reaction when using methanol as a mobile-phase component results in a “moving target,” which can, in turn, result in missing a 1D peak that then never appears on the second dimension chromatogram (8).
In this work, we revisited the topic of esterification with two objectives: 1) to determine the extent of the effect of losing acid content from the mobile phase on the retention and selectivity of different analytes; and 2) to develop a mitigation strategy for this problem.
Esterification of Formic Acid, and Its Effects on UV Detection Baselines
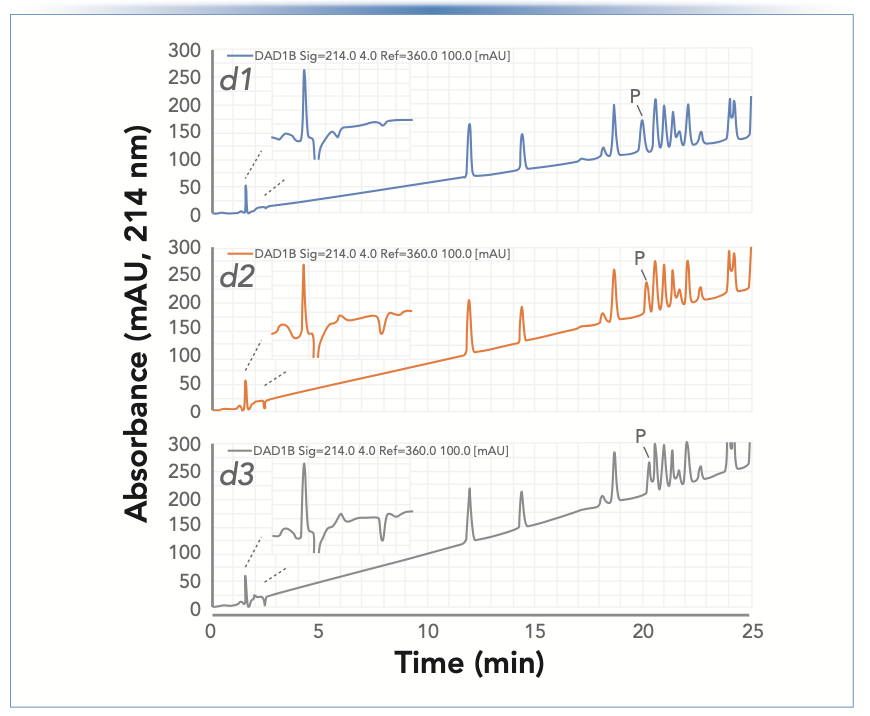
Figure 1 shows UV detection chromatograms from RP separations of a sample containing a low molecular weight, and ionizable compounds acquired over the course of three consecutive days (d1 to d3). The method used gradient elution from 20 to 70% B (B = 0.1% FA in methanol). Both the A and B solvents were freshly prepared on the first day. We see that the slope of the baseline at 214 nm continually increased over the course of three days, as did the magnitude of a negative peak shortly after a positive system peak. Retention times were stable over the three days, with the exception of the compound marked with P, which shifted towards later retention times from day 1 to day 3. The changes in the early part of each chromatogram were quite interesting. Along with a system peak (approximately 1.5 min), which we assume occurs because of a mechanism described previously (9), we believe the negative peak at approximately 2.5 min is because of the methyl formate produced in the B solvent (0.1% FA in methanol) since the moment the acid was added on day 1. As the esterification reaction proceeds, the amount of ester adsorbed onto the column in the starting mobile phase of the gradient (that is, 20% methanol) increases. At the same time, the UV absorption background caused by the ester increases. The injected plug of strong solvent (acetonitrile) in the sample matrix leads to desorption of the ester from the stationary phase and a local decrease in the ester content on the column because the sample itself does not contain the ester. These vacancies are subsequently replenished by methyl formate as it enters the column with the mobile phase. The larger amount of the ester needed to re-establish the mobile-stationary phase equilibrium in the column, and the higher background absorbance (which is usually not realized by an operator, because the detector is “zeroed” or “balanced” at the initial conditions in the run) lead to an increasingly large negative peak at the retention time of the ester, when the depletion zone arrives at the detector.

 and diluted 1:10 with acetonitrile using a Zorbax SB-C18 column (150 mm x 4.6 mm i.d., 5 μm). For the LC system, solvent A contained 0.1% FA in water; solvent B contained 0.1% FA in methanol; the gradient was 20–70% B over 25 min; the flow rate was 1 mL/min; the injection volume was 1 μL; the column temperature was 40 °C; and the detection by UV absorbance was at 214 nm.")
FIGURE 1: Separation of the 2D-LC starter sample over the course of three consecutive days (labeled d1 to d3) and diluted 1:10 with acetonitrile using a Zorbax SB-C18 column (150 mm x 4.6 mm i.d., 5 μm). For the LC system, solvent A contained 0.1% FA in water; solvent B contained 0.1% FA in methanol; the gradient was 20–70% B over 25 min; the flow rate was 1 mL/min; the injection volume was 1 μL; the column temperature was 40 °C; and the detection by UV absorbance was at 214 nm.
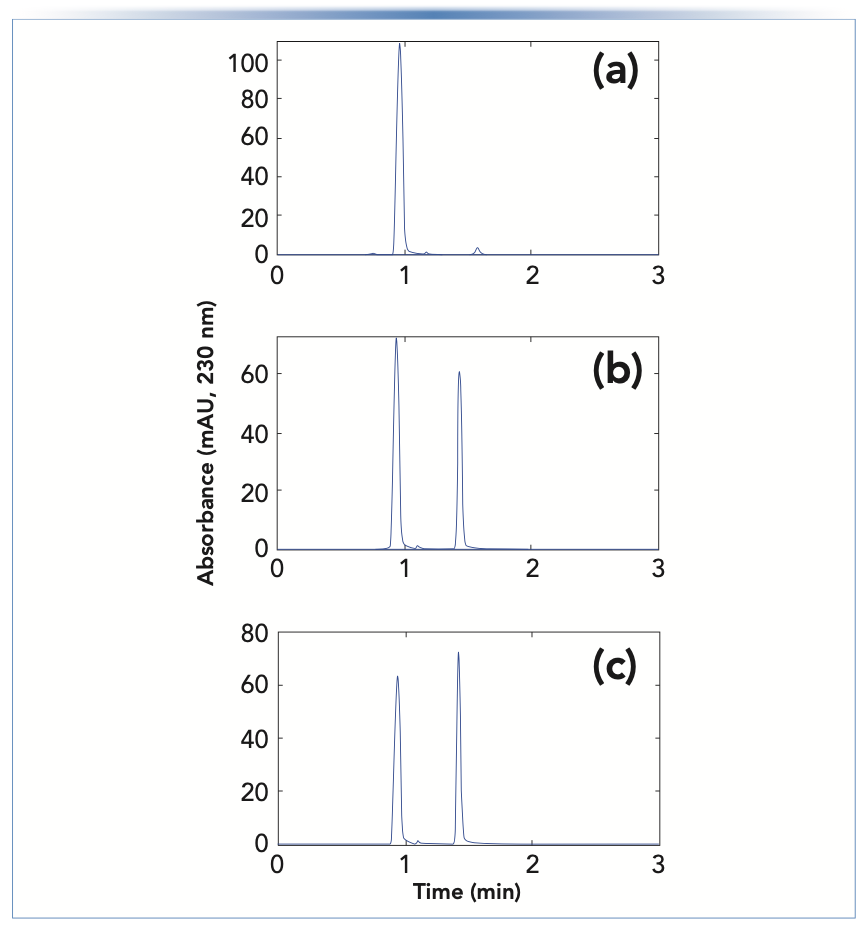
To confirm the esterification reaction in our hands, a mixture of 10 μL concentrated FA and 990 μL methanol was added to a vial, and injections of the mixture were made over the course of 24 h. A RPLC method was used with a mobile phase of 2:98 acetonitrile:water; the results are shown in Figure 2. After 10 min of reaction time, most of the observed peak area was because of FA (that is, the peak at approximately 1 min). However, there was also evidence of methyl formate eluting at 1.6 min, which was confirmed by spiking the sample with methyl formate (data not shown). Over time, the area of the ester peak at 1.6 min increased, with a concomitant decrease in the area of the peak because of the acid.

FIGURE 2: Separation of formic acid and methyl formate using a Poroshell 120 EC-C18 column (100 mm x 3.0 mm i.d., 2.7 μm). Chromatographic conditions: mobile phase, 2:98 acetonitrile:water; flow rate, 0.5 mL/min, column temperature, 35 °C; and detection by UV absorbance at 230 nm. Injections were made after (a) 10 min, (b) 14 h, or (c) 24 h.
Rationalizing the Effect of Formic Acid Loss on Retention of Amines
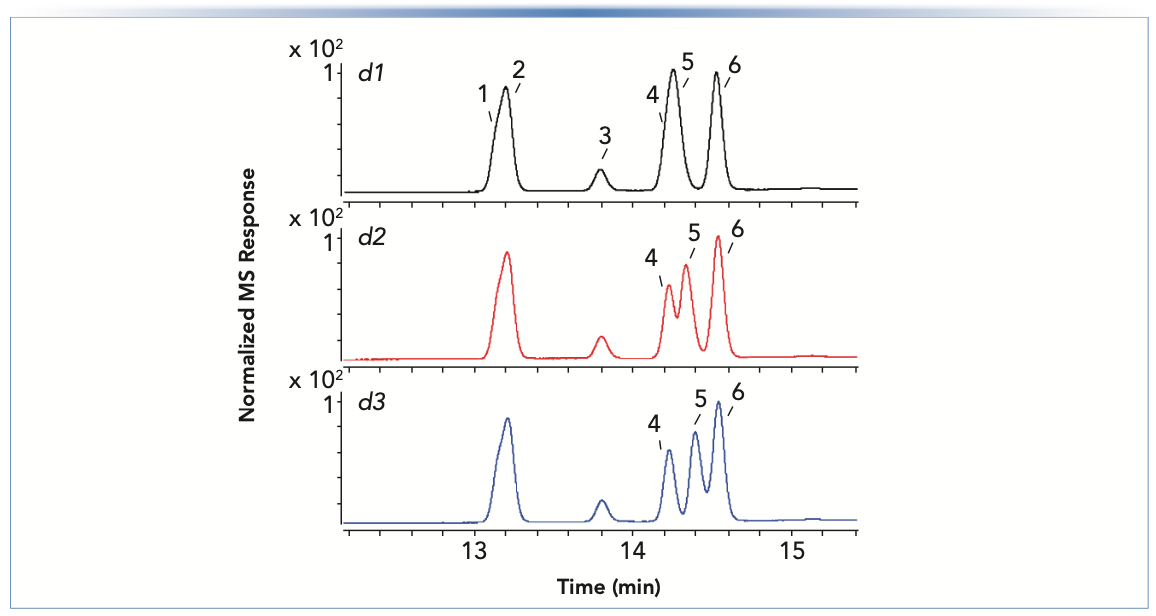
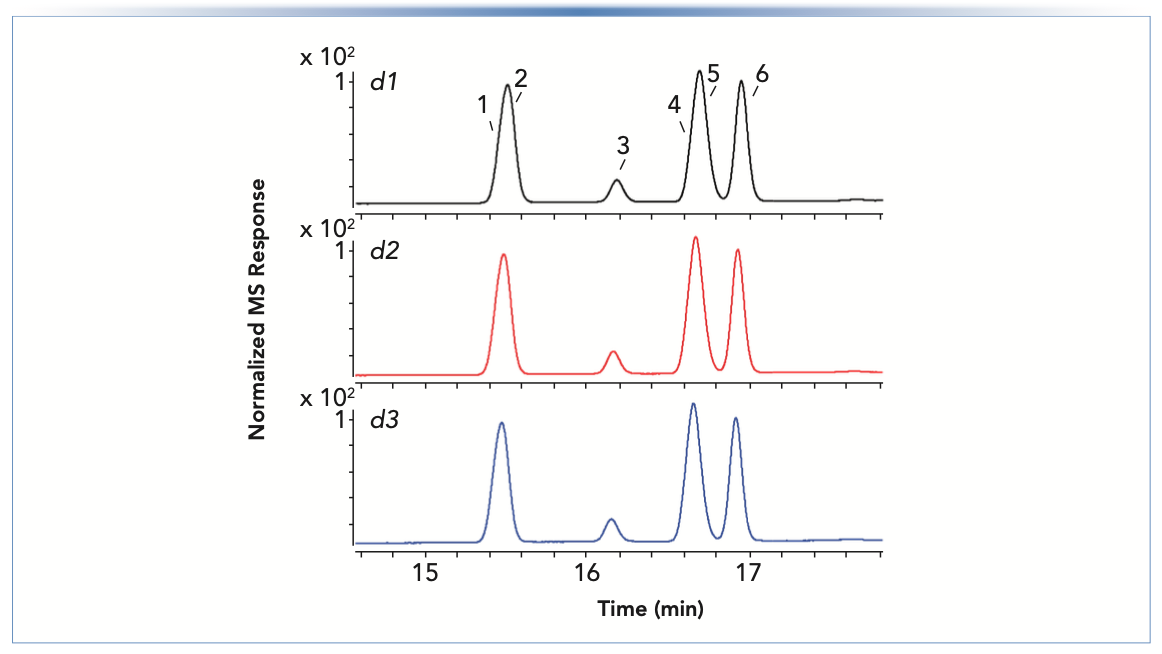
To understand why the retention of compound P increased over time while the other components of the sample were not affected (Figure 1), we added a mass spectrometric (MS) detector to identify the compounds. Figure 3 shows excerpts from RP separations of the 16-component sample acquired over the course of three days. Conditions were similar to those used in Figure 1, but adjusted to accommodate the requirements of the MS detector. Six compounds were confirmed by MS within the range shown: 1) chlortoluron (pKa, 14.4); 2) metobromuron (pKa, 12.8); 3) atrazine (pKa, 1.6); 4) methabenthiazuron (pKa, 12.8); 5) prometryn (pKa, 4.1); and 6) diuron (pKa, 13.6). Interestingly, the resolution of peaks 4 and 5 improved on days 2 and 3; however, these significant changes in retention and resolution under apparently identical conditions are not acceptable for routine use of such a method. Prometryn (peak 5 in Figure 3) has a pKa value of 4.1 in an aqueous solution at 20 °C (10). The pH of the mobile phase A solvent was 2.8 at room temperature. Both these values are subject to change upon the addition of methanol to the mobile phase during the gradient, with the column temperature set to 40 °C (relative to room temperature). It is likely that the mobile-phase pH and the pKa of prometryn approach each other under these conditions (11). Thus, a small increase in mobile-phase pH because of the loss of FA in the solution pushes the prometryn to a lower degree of ionization (that is, a higher ratio of neutral free amine to protonated, positively charged amine). Because the neutral, free amine is less hydrophilic, the observed retention time increases. The compounds (1, 2, 4, and 6)with a pKa far above the pH of FA are not affected because subtle changes in pKa and mobile-phase pH do not change their ionization state markedly. On the other hand, the retention of atrazine, whose pKa values lies within the region pH (FA) ±2 pH units, is only slightly affected, which shows that pH effects can be analyte specific.

FIGURE 3: Separations of the 2D-LC starter sample, diluted 1:100 with the initial mobile phase, over the course of three consecutive days (labeled d1 to d3), using a Zorbax SB-C18 column (100 mm x 2.1 mm i.d., 1.8 μm). Conditions are as in Figure 1, except that the solvent gradient was 25–87.5% B over 25 min with solvent B of 0.1% FA in methanol:water (80:20), and the flow rate was 0.4 mL/min. A triple quadrupole MS instrument was used in positive mode.
Esterification Occurs With Trifluoroacetic Acid in Methanol, Too
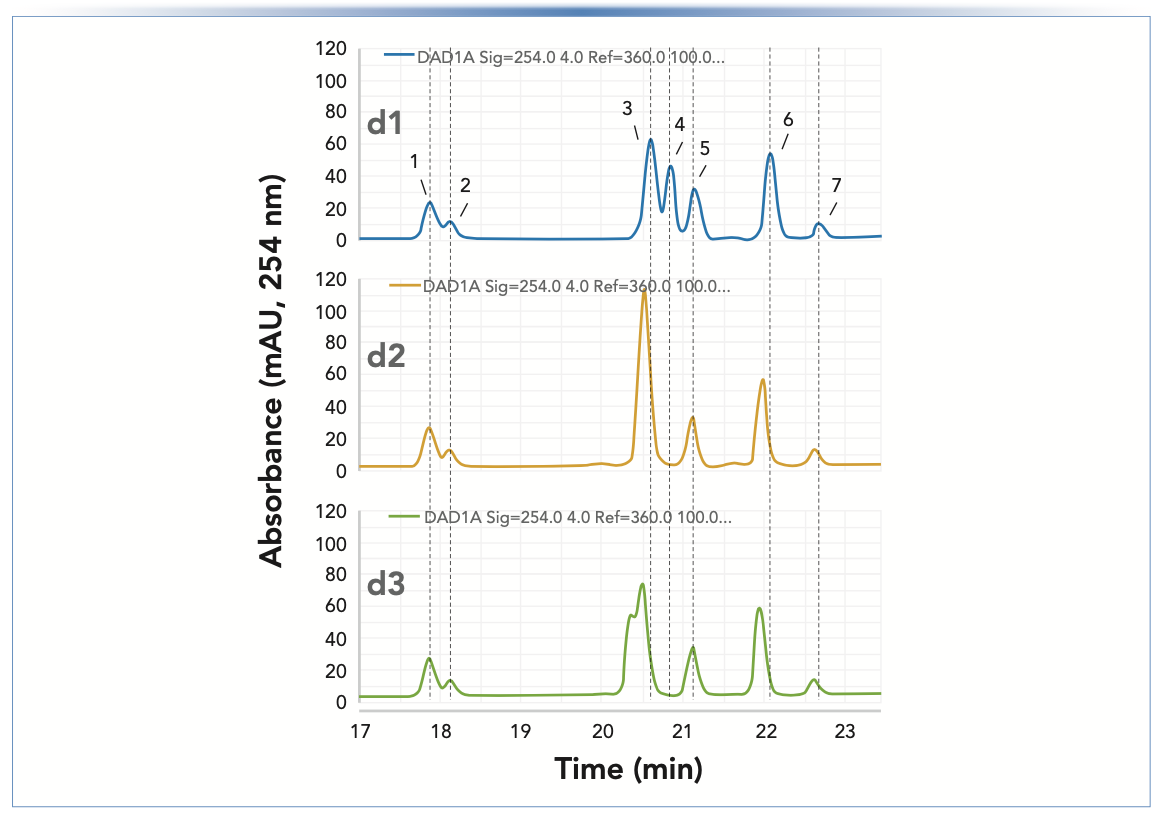
In a further study of the effects of esterification on LC separations, the A and B solvents used in Figure 1 were replaced by 0.1% TFA in water and 0.1% TFA in methanol, respectively. The pH of the solution of 0.1% TFA in water was 1.9; thus, any effects on the retention of prometryn (pKa ~ 4.1) caused by slight shifts in the mobile-phase pH (as described above) should be at least reduced. Figure 4 shows chromatograms obtained under the same conditions as those in Figure 1, except for the replacement of FA with TFA. As in Figure 1, the retention times of some peaks are not affected by the loss of mobile-phase acidity over the course of the three days (for example, peaks 1, 2, and 5). However, in contrast to the results obtained with FA, the retention times of some compounds decreased with TFA, and the magnitude of these decreases was smaller than the magnitude of the increase observed with FA. For instance, the retention times of peaks 3, 4, 6, and 7 all decreased, with the most pronounced change observed for peak 4. On day 1, peak 4 eluted after peak 3, but by day 3, the elution order had completely reversed. These results could be related to the ion pairing ability of TFA. When TFA is lost from the solution because of esterification, there is less TFA available for ion pairing with the analytes, rendering them less hydrophobic, and thus less retained under RP conditions.

FIGURE 4: Separations of the 2D-LC standard samples, diluted 1:100 with the initial mobile phase, over the course of three consecutive days (labeled d1 to d3), using a Zorbax SB-C18 column (150 mm x 4.6 mm i.d., 5 μm). Conditions are as in Figure 1, except that the A and B solvents were replaced by 0.1% TFA acid and 0.1% TFA acid in methanol, respectively, and the UV detection wavelength was 254 nm.
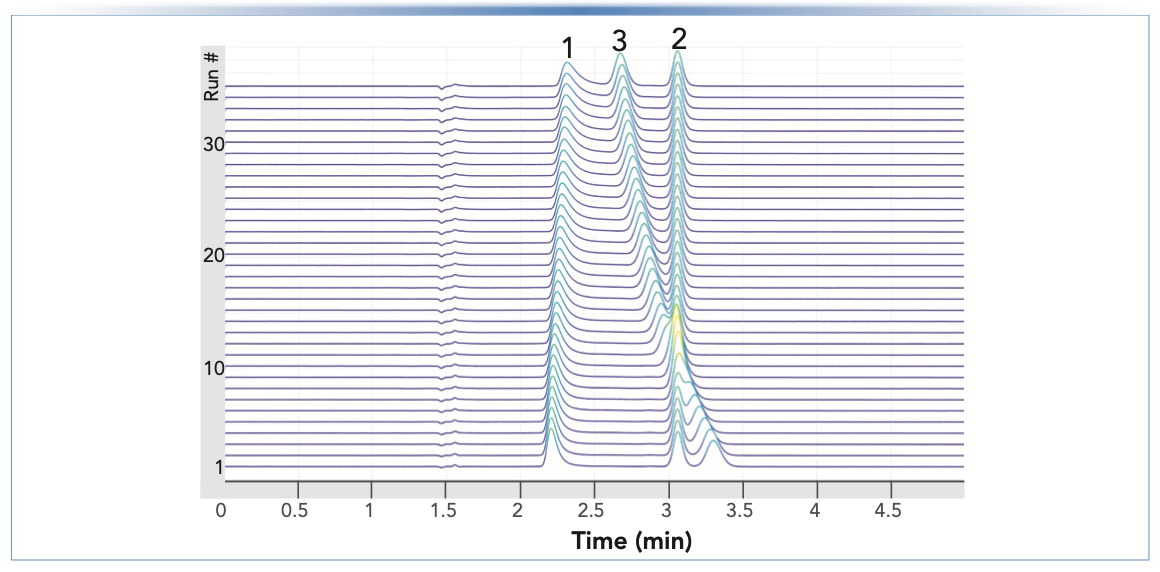
A final, remarkable example of the effect of esterification in acidified methanol is shown in Figure 5. In this case, a mixture of 2,6-dihydroxybenzoic acid (peak 1), salicylic acid (peak 2), and chlorhexidine (peak 3) was injected repeatedly over a period of three days. The reduction of TFA in the solution is accompanied by drift of the chlorhexidine peak towards shorter retention times (from 3.3 to 2.7 min). Chlorhexidine is a polyguanidine that is polycationic under most conditions and can form ion pairs with TFA, which may explain its extreme sensitivity to the effects of esterification chemistry.

FIGURE 5: Separation of a sample containing (1) 2,6-dihydroxybenzoic acid, (2) salicylic acid, and (3) chlorhexidine over three days (Run #1–35), using a Zorbax SB-C18 column (150 mm x 4.6 mm i.d., 5 μm). Chromatographic conditions: solvent A, 0.1% TFA acid in water; solvent B, 0.1% TFA acid in methanol; isocratic elution at 65% B; flow rate, 1 mL/ min; injection volume, 1 μL; column temperature, 25 °C; and detection by UV absorbance at 214 nm. The time interval between sample injections was approximately 2 h.
Solutions to the Problem
From the examples discussed in this installment, it is clear that esterification in acidified methanol can jeopardize the robustness of RPLC separations of small molecules, and, as already described by Przybytek and coworkers, chromatographically relevant levels of esterification of methanol occur even in presence of a large fraction of water (7). To avoid the effects of esterification on the separations, the methanol and acid must be kept apart until immediately before use, which could be achieved in at least two ways.
When using binary HPLC pumps, we have used 0.2% FA in water as solvent A and no acid in solvent B (data not shown). Though this approach provided retention stability, the drawback is that the FA concentration in the mobile phase changes as the gradient elution proceeds, which could cause a significant baseline slope when using UV detection. Alternatively, a quaternary pump could be used to blend the acid into the gradient as it proceeds. Figure 6 shows the same excerpt as in Figure 3, but from separations where a quaternary pump was used to deliver the mobile phase using the following solutions connected to three channels of the pump: a) water; b) methanol; and c) 0.5% FA in water. For the gradient elution program used for this method, the pump was programmed to deliver 20% of the flow from channel C while adjusting the flows from A and B to establish the methanol:water ratio. This approach yielded a 0.1% FA concentration in the mobile phase throughout the gradient, while keeping the methanol and acid apart until the blending point. A comparison of chromatograms obtained at day 1 (d1) in Figure 3 and 6 shows that the elution patterns are extremely similar. The retention times in Figure 6 are generally longer because of the larger gradient delay volume of the quaternary pump compared to a binary pump. However, in contrast to Figure 3, the pattern in the chromatograms of Figure 6 do not change markedly over the course of the three days of measurement.

FIGURE 6: Separation of the 2D-LC standard sample over the course of three consecutive days (labeled d1 to d3) that was diluted 1:100 with the initial mobile phase, using a Zorbax SB-C18 column (100 mm x 2.1 mm i.d., 1.8 μm). Conditions were the same as in Figure 1, except that the binary pump was replaced by a quaternary pump and the FA (0.5% FA in water) was blended into the gradient online to provide a constant concentration of 0.1% FA throughout the gradient. The flow rate was 0.4 mL/min.
Summary
We have demonstrated that the use of methanol acidified with FA or TFA as mobile-phase solvents can be problematic in RPLC separations of low molecular weight compounds. Chromatographically relevant levels of esterification of the methanol can lead to baseline artifacts and changes in both retention and selectivity. Although some analytes are insensitive to the depletion of acid from the methanol-containing solvent, retention of other compounds can change dramatically, resulting in poor reproducibility and even elution order reversal, which compromises method robustness. These problems can be avoided by adding acid to only the aqueous part of the mobile phase (for example, when using a binary pump) or when the acidified mobile phase is prepared “in situ” by online blending. This dramatically improves the repeatability of baselines, retention times, and selectivity on the timescale of days.
References
(1) L.R. Snyder, J.J. Kirkland, and J.W. Dolan, in Introduction to Modern Liquid Chromatography (Wiley, Hoboken, NJ, 3rd ed., 2010), pp. 303–360.
(2) D.V. McCalley, LCGC North Am. 17, 440–456 (1999).
(3) D.V. McCalley, J. Chromatogr. A. 987, 17–28 (2003). https://doi.org/10.1016/S0021-9673(02)01812-5.
(4) L.R. Snyder, in Practical HPLC Method Development (Wiley, Hoboken, NJ, 2nd ed., 1997), pp. 226–227.
(5) K. Croes, A. Steffens, D.H. Marchand, and L.R. Snyder, J. Chromatogr. A. 1098, 123–130 (2005). https://doi.org/10.1016/j.chroma.2005.08.090.
(6) C.G. Huber and A. Premstaller, J. Chromatography A. 849, 161–173 (1999). https://doi.org/10.1016/S0021-9673(99)00532-4.
(7) N.M. Fox, A.R. Kemperman, S.M. Lorenz, K.A.J. Snoble, and J.T. Przybytek, LCGC North Am. 26, 946–950 (2008).
(8) P. Petersson, LCGC North Am. 37, 740–746 (2019).
(9) K. Choikhet, B. Glatz, and G. Rozing, LCGC Europe 16(2), 2–9 (2003).
(10) National Library of Medicine, Prometryn. https://pubchem.ncbi.nlm.nih.gov/compound/Prometryn (accessed August 2022).
(11) S.M.C. Buckenmaier, D.V. McCalley, and M.R. Euerby, J. Chromatogr. A. 1026, 251–259 (2004). https://doi.org/10.1016/j.chroma.2003.11.007.
ABOUT THE CO-AUTHORS

Benedikt Metzger is a R&D scientist at Agilent Technologies, Germany. His focus is liquid chromatography and verification of instruments. He holds a M.Sc. in Chemistry from the Karlsruher Institute of Technology.


Konstantin Shoykhet is a principal R&D scientist at Agilent Technologies, Waldbronn, Germany, focusing on liquid chromatography and solvent delivery. He holds a Ph.D. in Chemistry from the University of the Saarland in the field of liquid chromatography and capillary electrophoresis.


Stephan Buckenmaier is a Principal Research Scientist in the Liquid Phase Separation Division of Agilent Technologies in Waldbronn, Germany. His primary focus is on the development of 2D-LC-MS workflow solutions and collaborations with academic institutions and external companies.

ABOUT THE COLUMN EDITOR

Dwight R. Stoll is the editor of “LC Troubleshooting.” Stoll is a professor and the co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 75 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC’s editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com



















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



