Analysis of Coumarin in Tobacco, Smokeless Tobacco Products, and Electronic Cigarette Liquids by Isotope Dilution LC–MS/MS
This article describes a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method for the analysis of coumarin in various tobacco matrices and electronic cigarette (E-cig) liquids, and highlights the importance of evaluating different MS/MS transitions of an analyte in complex sample matrices to overcome matrix effects. Matrix interfering components were separated from analyte using a C18 ultrahigh-pressure liquid chromatography (UHPLC) column with a larger inner diameter (3.0 mm, or 4.6 mm). Matrix suppressions on analyte responses were corrected by isotope dilution. Four different MS/MS transitions of coumarin were studied in each sample matrix to select a suitable MS/MS transition for analyte quantification based on matrix effects on each MS/MS transition. The method was validated using different tobacco matrices and E-cig liquids.
Coumarin (1,2-benzopyrone) occurs as a natural component in some plants, and is found as a flavoring ingredient in some foods, tobaccos, and cosmetic products. Coumarin has also been identified and determined as a natural constituent in different types of tobaccos (1). The high content of coumarin in some foods, tobacco products, and other consumer products has received considerable attention due to its hepatotoxic effects found in animal experiments (2). The European food safety authorities have set a maximum limit of 2 mg/kg for foods and beverages in general, and a maximum level of 10 mg/L for alcoholic beverages (3). Coumarin has been banned as a flavor additive in food and other products in the United States (4), and has been included by the U.S. Food and Drug Administration (FDA) on the established list as a harmful and potentially harmful constituent in tobacco and smokeless tobacco products (5).
Electronic cigarettes (E-cigs), also known as vaporizers or vaping products, are battery-powered devices to heat liquid-based nicotine into an inhalable vapor. E-cigs have been widely marketed as a safer alternative to traditional cigarettes, because they can quell smokers’ urges for nicotine without using cancer-causing tobacco. However, whether E-cig has less risk or more risk is still debatable due to the devices’ high nicotine content and other chemicals and additives in the liquids. E-cigs are by far the most popular tobacco product among teens, according to the 2017 national youth tobacco survey (6). The teens are attracted to vaping by the various flavors in the E-cig liquids. The U.S. FDA is weighing a ban on most flavored E-cigs following a recent national outbreak of E-cig or vaping product use-associated lung injury (EVALI), which is linked to vitamin E acetate used as an additive in some vaping products (7,8).
Although different analytical methods have been used for the determination of coumarin in plant extracts, foods, and fragrance products, the most widely used method in the past was based on high pressure liquid chromatography-ultraviolet spectroscopy (HPLC-UV) with sample clean up and concentrations (9). However, HPLC-UV method suffered from its drawbacks of low selectivity and sensitivity. Because of the low selectivity, the method could easily give false positive results for coumarin due to matrix interfering components, especially for complex sample matrices such as food and tobacco samples, and, therefore, it is required to use a longer analytical column and take longer runtime to separate coumarin from sample matrix components. Due to the low sensitivity of the method, extensive sample clean up and analyte concentration steps are often needed to achieve good separation and sensitive response for coumarin analysis. The gas chromatography-mass spectrometry (GC–MS) method is more selective than the HPLC-UV method and has been used for coumarin analysis in tobacco samples (1,10,11). But it still needs sample clean up and analyte concentration to achieve good sensitivity. Recently, the liquid chromatography-tandem mass spectrometry (LC–MS/MS) method has been developed for coumarin analysis in food samples, and demonstrated much higher sensitivity and selectivity with simpler sample preparation (12–16). In this study, a fast, sensitive, selective, and robust LC–MS/MS method has been developed for the analysis of coumarin in various tobacco sample matrices. To obtain accurate results from complex tobacco sample matrices, several reference tobacco samples were used during method validation and multiple MS/MS transitions were evaluated for coumarin identification and quantification. Compared to the GC–MS method, this LC–MS/MS method is much simpler (with no need for sample clean up and analyte concentration), faster, more selective, and sensitive. In addition, using a stable isotope labeled internal standard, this method is more accurate and robust and can be easily applied to coumarin analysis in tobacco, smokeless tobacco products (STP), and E-cig liquid samples in a routine testing laboratory.
Experimental
Chemicals and Materials
Coumarin (≥99% in purity) was obtained from Sigma-Aldrich and deuterium labelled coumarin-5,6,7,8-d4 (98% in purity) was obtained from Toronto Research Chemicals. LC–MS grade methanol, water, formic acid, and other chemicals were obtained from Sigma-Aldrich. The Kentucky reference cigarette tobacco (KY3R4F) was obtained from the Tobacco and Health Research Institute at University of Kentucky, in Lexington, Kentucky. Smokeless reference tobacco products (1S2 dry snuff and 2S3 moist snuff) and the Cooperation Centre for Scientific Research Relative to Tobacco (CORESTA) recommended reference smokeless tobacco products (CRP) were provided (free of charge) by the Department of Crop Science at North Carolina State University, in Raleigh, North Carolina. Two batches of CRP reference samples were obtained; one batch of samples was produced in 2009 and labeled as: CRP1 (snus), CRP2 (moist snuff), CRP3 (dry snuff) and CRP4 (chewing tobacco), and the other batch was produced in 2016 and labeled as: CRP1.1 (snus), CRP2.1 (moist snuff), CRP3.1 (dry snuff) and CRP4.1 (chewing tobacco). Other testing samples, including five brands of cigarettes and five brands of mini-cigar samples, as well as six brands of E-liquid samples, were obtained from a local store in Toronto, Canada.
Standard Preparation
The primary coumarin standard solution (10 mg/mL) and internal standard (IS) coumarin-d4 solution (1 mg/mL) were prepared in methanol, separately. The secondary coumarin standard solution (10 µg/mL) and internal standard solution (IS spiking solution, 10 µg/mL) were prepared separately by diluting their primary standard solutions with 50% methanol solution (in LC–MS grade water, v/v). A tertiary coumarin standard (1.0 µg/mL) was prepared by diluting the secondary solution with 50% methanol solution. Twelve levels of calibration standards containing coumarin at 0.05, 0.1, 0.5, 1, 5, 10, 20, 50, 100, 200, 500, and 1000 ng/mL were prepared from the secondary and tertiary standard solutions by dilutions with the 50% methanol solution. Each calibration standard contained 100 ng/mL internal standard. Two standard zero solutions were also prepared. Standard 01 was prepared by adding 50% of methanol solution directly into an auto sampler vial to check the background and potential contamination from the vials, and standard 02 contains only the 100 ng/mL of IS, which was prepared to check the isotope purity of the IS.
Tobacco and Smokeless Tobacco Sample Preparation
A 1.0 g sample of the homogenized tobacco product from a freshly opened source was spiked with 200 µL of the IS spiking solution, and then extracted with 20 mL of 50% ethanol solution (in Type I water, 1:1 in v/v) in a 50 mL flask and agitated for 30 min on a shaker. The sample solution was then filtered through a 0.22µm syringe filter into an 8 mL storage vial. A portion of the filtered solution was analyzed directly by the LC–MS/MS method after appropriate dilution with 50% methanol solution.
E-cig Liquid Sample Preparation
A 1.0 g sample of the E-cig liquid sample from a freshly opened source was spiked with 100 µL of the IS spiking solution and then diluted with 10 mL of 50% methanol solution in a 50 mL centrifuge tube and agitated for 10 min on a shaker. The sample solution was analyzed directly by the LC–MS/MS method without further sample treatment.
Quality Control Sample Preparation
To test possible interference or contamination from reagents or materials used and from the sample preparation processes, a laboratory reagent blank (LRB) was prepared per work shift by following the same procedures as for tobacco or E-cig liquid sample preparation described above, without adding tobacco or E-cig liquid sample. To study possible analyte loss or contamination during sample preparations, a Laboratory Fortified Blank (LFB) sample was prepared per work shift by following the same tobacco or E-cig liquid sample preparation procedures as described above, by spiking a known amount of analyte solution. During method validation, LFB samples were prepared by spiking the analyte in three different concentration levels, as shown in Table I, and three replicates of the LFB samples at each level were prepared. To evaluate sample matrix effects andanalyte recovery from sample matrix, a Laboratory Fortified Matrix sample (LFM) was prepared per work shift by following the same tobacco or E-cig liquid sample preparation procedures as described above, using 1.0 gram of a reference tobacco or E-cig liquid sample spiked with a known amount of analyte. The percent recovery is calculated by comparing the difference of the spiked (LFM sample) and non-spiked sample results and the expected (spiked) value. During method validation, the LFM samples were prepared using seven different reference tobacco sample matrices (CRP1.1, CRP2.1, CRP3.1, CRP4.1, 1S2, 2S3 and KY3R4F) and a mini cigar tobacco sample, and different concentrations of analyte were spiked onto each tobacco sample matrix as illustrated in Table I. The LFM samples for E-cig liquids were prepared using two different E-cig liquid sample matrices (with different nicotine contents and flavor components), and three different concentrations of analyte were spiked onto each sample matrix, as shown in Table I.



Analytical Conditions
Chromatographic separation of coumarin from matrix interfering components was conducted using an ultrahigh-pressure liquid chromatography (UHPLC) system (PerkinElmer LX-50) and coumarin detection was achieved using a triple quadrupole mass spectrometer (PerkinElmer QSight 220). Two LC–MS/MS systems were used for method robustness validation. Several available UHPLC columns were tested initially; including Phenomenex Kinetex PFP, C8, C18 and XB-C18 columns (2.6 µm, 100 × 4.6 mm), Phenomenex Kinetex C18 column (2.6 µm, 100 × 2.1mm), PerkinElmer Quasa SPP Pesticides column (C18, 2.7µm, 100 × 2.1mm and 100 × 4.6 mm) and PerkinElmer Brownlee SPP C18 (2.7µm, 100 × 2.1mm and 100 × 3.0 mm). The C8 and C18 columns with larger inner diameter (id) such as 3.0 mm or 4.6 mm provided good separation of coumarin from matrix interferences and thus were used during method robustness validation. Mobile phases were water (A) and methanol (B). LC separation was achieved using an isocratic elution of 60% mobile phase B for 6 min at a flow rate of 0.4 mL/min, followed by a gradient to 100% mobile phase B at 6.5 min to wash and clean the column. Finally, the mobile phases were returned to the initial composition at 8 min and then kept under the initial conditions for 4 min for column equilibration. The total run time for each sample including equilibration time was 12 min. Column temperature is 30 °C, and the injection volume is 5 mL. The auto sampler was controlled at 5 °C. Mass detection conditions were as follows: ionization mode: positive ESI/MRM, ion spray voltage: 2000 V; ion source temperature: 400 °C, HSID temperature: 320 °C, drying gas: 120 L/h, nebulizer gas: 400 L/h. Compound-dependent parameters, such as collision energies (CE), entrance voltages (EV), and the lens voltages (CCL2), were optimized by infusion of standards and use of the software. Four different fragment transitions (MS/MS) were monitored for coumarin under multiple reaction monitoring (MRM). The following optimized conditions were used for all the four transitions: dwell time, 85 ms; EV, 20 V; and CCL2, -45 V. The following collision energy (CE) was used for each transition, respectively: -33 eV for the first coumarin MS/MS transition (147.1/91.1) and coumarin-d4 (MS/MS: 151.1/95.1); -25 eV for the second coumarin MS/MS (147.1/103.1) and coumarin-d4 (MS/MS: 151.1/107.1); -48 eV for the third coumarin MS/MS (147.1/65.1) and coumarin-d4(MS/MS: 151.1/68.2); and -36 eV for the fourth coumarin MS/MS (147.1/77.1) and coumarin-d4 (MS/MS: 151.1/80.2). Quantification of coumarin was performed using the ratio of peak area of coumarin to that of internal standard coumarin-d4.
Results and Discussion
LC–MS/MS Method Optimization
For mass detection of coumarin, both positive and negative electrospray ionization (ESI) modes were evaluated initially. The results showed that the analyte gave better sensitivity and better signal to noise ratio under positive mode and therefore positive ESI detection was used in this study. For coumarin and its internal standard (IS) coumarin-d4, several product ions were generated at certain collision energies and thus multiple MS/MS transitions could be formed. The optimized MS/MS parameters were listed in the above experimental section in the order of signal intensity.
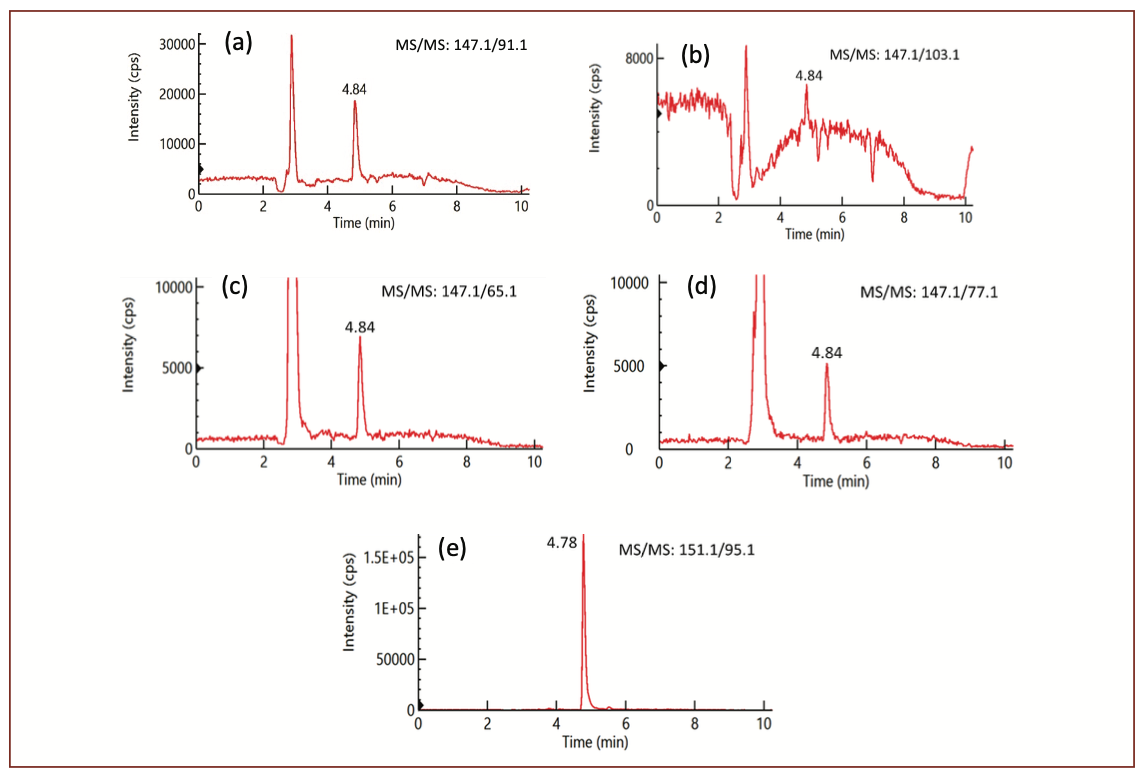
To separate coumarin from interfering components in different tobacco sample matrices, several reversed phase UHPLC columns available in our laboratory were evaluated. The Kinetex PFP (pentafluorophenyl propyl) column was confirmed not suitable for this study because very broad and splitting analyte peaks were found with this column. All the tested C8 and C18 columns, including Quasar SPP C18, Brownlee SPP C18, Kinetex C8, C18, and XB-C18 columns, could be applied to the analysis of coumarin. But it was found that the columns with smaller inner diameters (2.1 mm in this study) had difficulty to separate coumarin from interfering components, especially for the third MS/MS transition (147,1/65.1) chromatograms due to relatively stronger interfering component peaks at this transition as shown in Figure 1c. The C8 and C18 columns with larger column inner diameters (4.6 mm or 3.0 mm) provided much better separation efficiency, due to a higher column peak capacity, and therefore were used during method validation and final application. Mobile phase compositions of methanol/water and acetonitrile/water with and without acid were evaluated and it was found that methanol/water provided the highest analyte signal intensity. Figure 1 shows an example of separating coumarin peak from interfering components in a smokeless tobacco sample. Figure 2 gives an example of separating coumarin peak from interfering components in a E-cig liquid sample.

FIGURE 1: (a-e) LC–MS/MS chromatograms for the four MS/MS transitions of coumarin in a smokeless reference tobacco sample CRP3.1.: (a) MS/MS 147.1/91.1; (b) MS/MS 147.1/103.1; (c) MS/MS 147.1/65.1 and (d) MS/MS 147.1/77.1, and for its internal standard (e) MS/MS 151.1/95.1.


FIGURE 2: (a-e) LC–MS/MS chromatograms for the four MS/MS transitions of coumarin in an E-cig liquid sample: (a) MS/MS 147.1/91.1; (b) MS/MS 147.1/103.1; (c) MS/MS 147.1/65.1 and (d) MS/MS 147.1/77.1, and for its internal standard (e) MS/MS 151.1/95.1.
Extraction of Coumarin from Tobaccoand Smokeless Tobacco Products (STP)
The method extraction efficiency for the targeted analytes is one of the most important parameters that will influence the method’s precision, accuracy, and robustness. The extraction efficiency depends on the solubility of the analytes in a certain solvent or a mixture of solvents, the number of extraction cycles applied, the extraction time in each extraction cycle, extraction temperatures and extraction techniques used. Coumarin has been extracted from tobacco samples by solid-liquid extraction method with different solvents. Various techniques have been used to improve coumarin extraction efficiency, such as stirring the solution using a magnet stir bar or shaking it on a mechanical shaker (1,15), using temperature controlled ultrasonic bath (11,14) and microwaves-assisted solvent extraction (16). However, these studies have shown that there is no significant difference in extraction efficiency among the different extraction techniques used, and higher temperatures can lead to loss of coumarin (1). Therefore, the most convenient technique of shaking sample solutions on a mechanical shaker at room temperature was applied in this study. In addition, we found that more than 90% of coumarin could be extracted from the studied tobacco sample matrices by a single 30 min extraction cycle using a solvent mixture of ethanol/water (1:1 in v/v) (experimental data on extraction study are available upon request).
E-cig Liquids Sample Preparation
Since both coumarin and the E-cig liquid solvents (glycerin and propylene glycol) are soluble in water, methanol and the mixture of water and methanol, the solvent mixtures with different ratios of methanol to water were studied for their effect on coumarin determination from E-cig liquid samples. The results showed no significant differences between these solutions and therefore, a 50% methanol aqueous solution was finally used as diluent for coumarin analysis. The sample solutions were mixed on a Vertex Mixer at room temperature before LC–MS/MS analysis.
Sample Matrix Effects and Method’s Selectivity
Sample matrix effects have attracted great attention in LC–MS/MS method development and validations since they affect the data quality and method’s selectivity, sensitivity, and accuracy, especially for the analyses of analytes in complex sample matrices such as food and tobacco samples (17–21). In this work, three important parameters of sample matrix effects were studied:
Matrix interfering peaks. Several tobacco matrix peaks were observed from chromatograms of tobacco samples, which could interfere with coumarin peak and affect coumarin results if they were not separated from coumarin peak. As illustrated in Figure 1, these matrix peaks could be separated from coumarin using a high efficiency UHPLC column.
Signal suppressions. In previous publications (18–20), the author had studied in detail on tobacco matrix effects and found that tobacco matrix could cause significant analyte signal suppression. These suppression effects were further confirmed in this study by spiking the same amount of IS into pure sample matrices (such as LRB samples without tobacco) and into different tobacco matrices and then comparing the signal intensity of IS in tobacco samples with those in pure samples. The results showed that tobacco matrix effects led to 1.5 to 5 times decrease in IS signal depending on the sample matrices (data not shown, but available upon request). To overcome these suppression effects and achieve accurate results, we used stable isotope labeled IS in all samples to compensate for sample matrix effects.
Matrix effects on different MS/MS transitions of an analyte. Since different samples can have different matrix effects on coumarin, sample matrix effects were studied for each tobacco sample matrix in this work. In addition, four MS/MS transitions of coumarin and its internal standard in each sample were carefully examined and the results showed that matrix interfering peaks and ion suppression effects were different among the four different MS/MS transitions; some transitions showed many matrix peaks and suffered from heavy suppression effects (Figure 1c), while other transitions experienced less matrix effects (Figure 1b and Figure 2a). For example, although the highest intense peak (147.1/91.1 in this study) is usually used for analyte quantification to increase the sensitivity of a method, the second intense peak (147.1/103.1) of coumarin is a better choice for coumarin quantification in tobacco and STP samples due to less matrix interferences for this MS/MS transition as shown in Figure 1b. However, the results are different when analyzing electronic cigarette liquid samples. As illustrated in Figure 2, severe matrix effects and interfering peaks were observed for the second intense peak (147.1/103.1), while the highest intense peak (147.1/91.1) gave the best coumarin results due to less sample matrix effects (Figure 2a). In both cases, the IS peaks (Figure 1e and Figure 2e) had less matrix effects. Therefore, it is important and necessary to examine all MS/MS transitions for each sample matrix before analysis to select the most suitable transition for analyte quantification.
The method’s selectivity and analyte confirmation from samples can be evaluated by comparing the analyte retention time and mass spectrum information between reference standard and tobacco samples. According to the regulatory guidance on analytical method validation, at least two structurally specific MS/MS transitions should be used in a method (22,23). In this study, four MS/MS transitions were examined for each sample. As shown in Table II, using a reference smokeless tobacco sample CRP3.1 as an example, the peak area ratios of qualifier to quantifier ions for the tobacco sample are consistent with those of reference standard with variations less than 15%. Similar results were also obtained for other tobacco and STP samples (data not shown, but available upon request), demonstrating good purity of the coumarin peaks for all four MS/MS transitions and great selectivity of the method for coumarin analysis in tobacco samples. Alternatively, the selectivity can also be evaluated by comparing the coumarin results obtained by the calibration methods built from each of the four MS/MS transitions. If the coumarin results obtained from each of the four MS/MS transitions are consistent, it indicates that there are no interfering peaks in any of the four MS/MS transitions used and the method is selective for determining coumarin in the samples. However, if the coumarin result obtained from one MS/MS transition is significantly higher than those obtained from each of the other three MS/MS transitions, it indicates that there are impurity peaks that are coeluted with the coumarin peak in this transition. If the coumarin result obtained from one MS/MS transition is significantly lower than those obtained from each of the other three MS/MS transitions, it indicates that there are severe signal suppression effects in this transition. As shown in Tables II and III, the consistent coumarin results obtained by using any of the four MS/MS transitions as a quantifier for coumarin determination indicate that there are no interfering peaks in any of the four MS/MS transitions and the method is selective for determining coumarin from tobacco and STP samples. However, for E-cig liquid samples, as demonstrated in Table IV and Figure 2b, the second coumarin MS/MS transition (147.1/103.1) suffered from heavy matrix effects in this E-liquid sample, and thus led to much lower coumarin result and lower peak area ratio of qualifier to quantifier ions, while other three MS/MS transitions gave consistent results for both coumarin content and peak area ratio values between standard and sample. Thus, for E-cig liquid samples, the second MS/MS ion pairs should not be used for coumarin quantification. These results demonstrated the advantages of using multiple MS/MS transitions of an analyte in a LC–MS/MS method, which could help compounds confirmation and achieve more accurate results.






Method Validation
Several sets of calibration curves with concentrations ranging from 0.05 ng/mL to 1000 ng/mL were generated on separate days using each of the four MS/MS transitions. All the calibration curves showed good linearity with correlation coefficients (R2) greater than 0.999. Therefore, all the four MS transitions could be used for coumarin quantification if there are no interfering components in the peaks. The method’s limit of detection (LOD) and limit of quantification (LOQ) were determined based on signal to noise ratio (S/N = 3 for LOD and S/N = 10 for LOQ) of the quantifier ion peaks in sample matrices. Although very low LOD (0.02 ng/mL) and LOQ (0.05ng/mL) can be obtained in pure standard samples, the LOD and LOQ for real tobacco matrices are relatively higher due to heavy matrix suppression effects. The LOD and LOQ of the method for tobacco samples are 10 ng/g of coumarin/sample (corresponding to 0.5 ng/mL of coumarin in final solution) and 40 ng/g (which corresponds to 2.0 ng/mL of coumarin in final solution). Similarly, the LOD and LOQ of the method for E-cig liquid samples are 2 ng/g of coumarin/sample (corresponding to 0.2 ng/mL of coumarin in final solution) and 5 ng/g (which corresponds to 0.5 ng/mL of coumarin in final solution). As shown in Table I, no interference or contamination from reagents or glassware was observed in this study as demonstrated by the LRB sample results. Good recoveries were obtained for LFB samples, indicating no analyte loss or contamination during sample preparations. Method precision was assessed based on replicate analyses of a standard and reference tobacco samples (5 replicates) on three days. The precision was then calculated based on the coefficient of variation (RSD %) of the collected data. The RSDs were 2.6~5.8% for the standard and 3.8~10.9% for tobacco samples, respectively. Method accuracy assesses how close the experimental value is to the expected value. Method accuracy was evaluated by the recovery of known amount of analyte spiked to samples (LFM samples). As shown in Table I, the recoveries of coumarin from the spiked samples were between 84% and 120%, demonstrating good accuracy of the method. Method robustness was studied by slightly changing the experimental parameters such as the ratio of ethanol and water in extraction solution, mobile phase gradients and LC columns from different suppliers. No significant differences in method’s performance and analyte results were observed and thus confirmed the robustness of the method. In addition, the method was validated on two LC–MS/MS systems with equivalent results obtained.
Determination of Coumarin in Tobacco and STP Samples
The method was applied to the determination of coumarin in different tobacco and STP samples including five brands of commercial cigarette tobacco samples, five brands of mini-cigar tobacco samples and various reference tobacco and STP samples. Five replicates for each brand of sample were prepared and analyzed. Although no coumarin was found from the studied cigarette and cigar tobacco samples as well as some STP samples (CRP1, CRP1.1, CRP4, and CRP4.1), significant amount of coumarin was determined from four brands of STP reference products, as listed in Table III. It should be noticed that the coumarin amounts found in CRP2.1 and CRP3.1 (produced in 2016) are much higher compared to coumarin in CRP2 and CRP3 (produced in 2009), indicating the possible loss of coumarin during storage period. Therefore, it is important to measure coumarin from freshly opened samples as quickly as possible and take consideration of their storage conditions (temperature and time) when comparing the results of different or similar tobacco samples.
Determination of Coumarin in E-cig Liquid Samples
Six different brands of E-cig liquid samples were analyzed by the method and significant amount of coumarin (111 ng/g) was found in one of the samples.
Conclusions
The objective of this study is to develop a simple, fast, sensitive, selective, and robust analytical method for the determination of coumarin in various tobacco sample matrices. This goal was achieved by coupling UHPLC with tandem mass spectrometry. The selectivity and accuracy of the method could be improved significantly by using stable isotope internal standard and multiple MS/MS transitions of coumarin. The method was validated with reference tobacco samples and was applied to the analysis of coumarin in cigarette tobacco, cigar tobacco, smokeless tobacco and electronic cigarette liquid samples with good linearity, precision, and accuracy. Compared to the HPLC-UV methods, this LC–MS/MS method is more sensitive, selective, and accurate. Compared with the GC-MS methods, this method is simpler, faster, and without the need for sample cleanup and analyte concentration.
Acknowledgements
The authors wish to thank the Department of Crop Science at North Carolina State University (Raleigh, North Carolina) for providing us all the smokeless reference tobacco products free of charge.
References
(1) Christakopoulos, A.; Feldhusen, K.; Norin, H.; Palmqvist, A.; Wahlberg, I. Determination of Natural Levels of Coumarin in Different Types of Tobacco Using a Mass Fragmentographic Method. J. Agric. Food Chem. 1992, 40 (8), 1358–1361. DOI: 10.1021/jf00020a014
(2) Lake, B. G. Coumarin Metabolism, Toxicity, and Carcinogenicity: Relevance for Human Risk Assessment. Food Chem. Toxicol. 1999, 37 (4), 423–453. DOI: 10.1016/s0278-6915(99)00010-1
(3) European Council, Council Directive (EEC) No. 88/388 on the Approximation of the Laws of the Member States Relating to Flavourings for Use in Foodstuffs and to Source Materials for Their Production. Off. J. Europ. Comm. 1988, L184, 61–66.
(4) U.S. Food and Drug Administration (FDA), Title 21-Food and Drugs, Chapter 1, Part 3, 3.33 Status of Foods Containing Added Coumarin. Fed. Regist. 1954, 19 (44), 1239–1240. https://www.govinfo.gov/content/pkg/FR-1954-03-05/pdf/FR-1954-03-05.pdf
(5) Harmful and Potentially Harmful Constituents (HPHCs) in Tobacco Products and Tobacco Smoke: Established list. U.S. Food and Drug Administration (FDA), 2012. https://www.fda.gov/tobacco-products/rules-regulations-and-guidance/harmful-and-potentially-harmful-constituents-tobacco-products-and-tobacco-smoke-established-list
(6) Centers for Disease Control and Prevention (CDC, US) Press Release, “Youth Tobacco Use Drops During 2011-2017,” 2018. https://www.cdc.gov/media/releases/2018/p0607-youth-tobacco-use.html
(7) Centers for Diseases Control and Prevention (CDC, US), Vitamin E Acetate associated with Lung Injury (EVALI). 2020. https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html
(8) U.S. FDA. Enforcement Priorities for Electronic Nicotine Delivery Systems (ENDS) and Other Deemed Products on the Market Without Premarket Authorization. Guidance for Industry, 2020. https://www.fda.gov/media/133880/download
(9) Scotter, M. J.; Roberts, D. P. T.; Rees, G. O. Development and Single-Laboratory Validation of an HPLC Method for the Determination of Coumarin in Foodstuffs Using Internal Standardization and Solid-Phase Extraction Cleanup. Anal. Methods 2011, 3 (2), 414–419. DOI: 10.1039/C0AY00545B
(10) Stanfill, S. B.; Brown, C. R.; Yan, X.; Watson, C. H.; Ashley, D. L. Quantification of Flavor-Related Compounds in the Unburned Contents of Bidi and Clove Cigarettes. J. Agric. Food Chem. 2006, 54 (22), 8580–8588. DOI: 10.1021/jf060733o
(11) Li, P.; Zhu, X.; Hong, S.; Tian, Z.; Yang, J.Ultrasound-Assisted Extraction Followed by Dispersive Liquid–Liquid Microextraction Before Gas Chromatography-Mass Spectrometry for the Simultaneous Determination of Flavouring Compounds in Tobacco Additives. Anal. Methods 2012, 4, 995–1000. DOI: 10.1039/C2AY05866A
(12) De Jager, L. S.; Perfetti, G. A.; Diachenko, G. W. Comparison of Headspace-SPME-GC–MS and LC–MS for the Detection and Quantification of Coumarin, Vanillin, and Ethyl Vanillin in Vanilla Extract Products. Food Chem. 2008, 107 (4), 1701–1709. DOI: 10.1016/j.foodchem.2007.09.070
(13) De Jager, L. S.; Perfetti, G. A.; Diachenko, G. W. Determination of Cumarin, Vanillin, and Ethyl Vanillin in Vanilla Extract Products: Liquid Chromatography Mass Spectrometry Method Development and Validation Studies. J. Chromatogr. A 2007, 1145 (1–2), 83–88. DOI: 10.1016/j.chroma.2007.01.039
(14) Raters, M.; Matissek, R. Analysis of Coumarin in Various Foods Using Liquid Chromatography with Tandem Mass Spectrometric Detection. Eur. Food Res. Technol. 2008, 227, 637-642. DOI: 10.1007/s00217-007-0767-9
(15) Rychlik, M. Quantification of Free Coumarin and its Liberation from Glucosylated Precursors by Stable Isotope Dilution Assays Based on Liquid Chromatography-Tandem Mass Spectrometric Detection. J. Agric. Food Chem. 2008, 56 (3), 796–801. DOI: 10.1021/jf0728348
(16) Vierikova, M.; Germuska, R.; Lehotay, J. Determination of Coumarin in Food Using Ultra-Performance Liquid Chromatography–Electrospray-Tandem Mass Spectrometry. J. Liquid Chromatogr. & Rel. Technol. 2008, 32 (1), 95–105. DOI: 10.1080/10826070802548689
(17) Zhang, K.; Wong, J. W.; Safarpour, H.; Krynitsky, A. J. Important Considerations Regarding Matrix Effects When Developing Reliable Analytical Residue Methods Using Mass Spectrometry. LCGC North Am. 2017, 35 (7), 444–451. https://www.chromatographyonline.com/view/important-considerations-regarding-matrix-effects-when-developing-reliable-analytical-residue-method
(18) Wu, J.; Joza, P.; Sharifi, M.; Rickert, W.S.; Lauterbach, J. H. Quantitative Method for the Analysis of Tobacco-Specific Nitrosamines in Cigarette Tobacco and Mainstream Cigarette Smoke by Use of Isotope Dilution Liquid Chromatography Tandem Mass Spectrometry. Anal. Chem. 2008, 80 (4), 1341–345. DOI: 10.1021/ac702100c
(19) Wu, J. Analysis of Tobacco-Specific Nitrosamines in Cigarette Tobacco, Cigar Tobacco, and Smokeless Tobacco by Isotope Dilution LC–MS/MS. LCGC North Am. 2020, 38 (10), 559–568. https://www.chromatographyonline.com/view/analysis-of-tobacco-specific-nitrosamines-in-cigarette-tobacco-cigar-tobacco-and-smokeless-tobacco-by-isotope-dilution-lc-ms-ms
(20) Wu, J.; Rickert, W.S.; Andrew, M.; Joza, P. Determination of N-nitrososarcosine in Tobacco and Smokeless Tobacco Products Using Isotope Dilution Liquid Chromatography Tandem Mass Spectrometry. Anal. Methods 2012, 4, 3448–3452. DOI: 10.1039/C2AY25540E
(21) Yang, P.; Chang, J. S.; Wong, J. W.; Zhang, K.; Krynistsky, A. J.; Bromirski, M.; Wang, J. Effect of Sample Dilution on Matrix Effects in Pesticide Analysis of Several Matrices by Liquid Chromatography–High-Resolution Mass Spectrometry. J. Agric. Food Chem. 2015, 63 (21), 5169–5177. DOI: 10.1021/jf505168v
(22) U.S. Food and Drug Administration, Bioanalytical Method Validation Guidance for Industry, 2018, pp. 1–41. https://www.fda.gov/media/70858/download
(23) European Commission, 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results, Off. J. Eur. Communities. 2002, L221, 8–36.https://op.europa.eu/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858
About the Author
Jingcun Wu is with PerkinElmer Canada, in Woodbridge, Ontario, Canada. Direct correspondence to: jingcun.wu2@perkinelmer.com

















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.
, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

