Advances in the Analysis of Persistent Halogenated Organic Compounds
LCGC Europe
This article reviews advances in the analysis of persistent halogenated organic compounds over the last century.
An important goal in the field of analytical chemistry is to achieve continual improvement in the analysis of persistent toxic pollutants. Halogenated organic compounds represent an important group of pollutants. They are used in a wide variety of applications such as flame retardants, fire suppressants, heat-transfer agents, surfactants and pesticides, mainly because of their chemical inertness and stability. As a result of this stability, many of these halogenated organic compounds are persistent in the environment, toxic and bioaccumulative in the food chain, and are consequently associated with adverse effects on human health and the environment.



The Stockholm Convention on Persistent Organic Pollutants (POPs) (2001)1 is an international treaty that focuses on the elimination or reduction of a number of these compounds or groups of compounds, which include pesticides, industrial chemicals and unintentionally produced POPs. The list of the original twelve compounds or groups of compounds and second group of nine is summarized in Table 1, along with an additional group of three compounds currently being reviewed for addition. The persistence and toxicity of these compounds as well as many thousands of others2,3 has driven the discovery and development of new analytical equipment and techniques in the interest of protecting humans and wildlife. Sensitivity, selectivity, speed of analysis and cost (four key method attributes) need to be considered when selecting the most appropriate method of analysis. The proper extraction, preparation and instrumental techniques must be selected to ensure that the uncertainty of the technique meets the required data quality objectives and the method is fit for the purpose for which it was intended.4 Continual improvement should be the prime consideration of any analytical laboratory with the key method attributes optimized for the test at hand.


Table 1: Stockholm convention POPs.
Halogenated organics have been produced by humans intentionally and unintentionally for hundreds of years. The greatest challenge through the years has been finding analytical methods that are sensitive and selective enough to determine concentrations at levels low enough to protect humans and wildlife. The current standard for analysis of organohalogens is gas chromatography (GC) with an appropriate detector that meets the required selectivity [e.g., mass spectrometry (MS) or electron capture detection (ECD)]. Unfortunately, methods capable of determining concentrations to meet the above criteria have only been available for the last few decades.
Early Non-chromatographic Methods
Halogenated organic compounds such as polychlorinated biphenyls (PCBs)5 and polychlorinated naphthalenes (PCNs)6 have been used for more than 100 years. PCNs, first synthesized in 1833 were not used extensively until the early 1900s when they found applications as flame retardants in fabrics, dielectric fluids and anti-fungal agents for gas masks. Methods of analysis for these halogenated POPs at that time were not sensitive or selective. The Carius method (pre-1900)7 determined organic halides by boiling samples with nitric acid in the presence of silver nitrate followed by gravimetric determination of the silver halides that precipitate from solution. In the Stepanow8 or Bacon9 method (early 1900s) the organic halide was reacted with sodium in the presence of ethanol and the chloride was determined using a Volhard titration (silver nitrate was added in excess which reacts with the chloride produced). The excess silver was back titrated with thiocyanate in the presence of Fe3+, which forms a bright red complex with the thiocyanate when the excess silver was complexed. The Stepanow and Bacon titration methods reduced detection limits to the part-per-thousand range from the low percent levels previously achieved by the Carius method.
Occupational exposure of industrial workers to organohalogens accelerated the need for new analytical techniques. Chloracne, first reported in 1899, is a disease caused by extensive exposure to halogenated organics.10 Chloracne resulting from PCN contamination was first reported by Wauer in 1918.6 Factory workers producing and using Halowaxes and other PCN formulations also exhibited nausea, anorexia, vertigo, jaundice and death. Substitutes, such as PCBs, were considered less toxic and replaced PCNs in many applications. However, by 1937 it was recognized that PCBs also caused similar effects and that factory workers often exhibited chloracne as well as the other effects listed above for PCNs.11 During the early part of the previous century occupational exposure was common. Warren Crummett in Decades of Dioxin12 writes about his observations in 1943 on the first days at work as a chemist in a chemical plant saying: "I was scared. It appeared obvious to me that this is a high-risk place to work. Chlorine and nitrosyl chloride leaks into the plant occurred frequently. One day I unwittingly stepped into a pocket of such gas. The choking impact knocked me to the floor and I had to report to the health department for oxygen inhalation treatment. Almost all the procedures on this job were hazardous."
Dichlorodiphenyltrichloroethane (DDT) was first synthesized in 1874, but its insecticidal properties were not discovered until 1939. DDT was used to control mosquitoes for malaria and lice for typhus during the Second World War.13–15 The use of DDT and other organochlorine (OC) pesticides skyrocketed in 1940s and 50s to control pests and increase crop yields. During that time DDT was analysed using a colorimetric method developed by Schechter et al.16 where DDT and degradation products dichlorodiphenyldichloroethylene (DDE) and dichlorodiphenyldichloroethane (DDD) were subjected to fuming nitric acid to produce a tetranitro-DDT complex that is reacted with a sodium methylate-methanol reagent. The resulting compound can be determined with maximum adsorption at around 600 nm. Levels down to 10 μg (~10 ppm) could be detected.
The Advent of Gas Chromatography
Although not used for quantitative trace level analysis until the 1950s, the concept of chromatography had been reported many years earlier, by Tswett17 and Day18 who reported that dyes and crude oil fractions were selectively retained on solid substrates respectively. Other observations of selective gas adsorption and separation on carbon and silica gel19 were also reported in literature during the first half of the 20th century. In 1941, Martin and Synge,20 who performed research on liquid–liquid partitioning experiments, reported that, "the mobile phase need not be a liquid but may be a vapour," and that, "very refined separations of volatile substances should therefore be possible in a column in which permanent gas is made to flow over gel impregnated with a non-volatile solvent." The practical development of gas chromatography (GC) was not realized until 195221–23 when Martin and James were able to separate and quantify four organic acids on a 4 ft × 4 mm column packed with DC-silicone oil containing 10% stearic acid using N2 as the carrier gas. Figure 1 shows the first published gas chromatogram. The eluants were detected by titration with sodium hydroxide and phenol red indicator. The detection limit was 20 μg and the column had 700 plates with a peak capacity of 5 to 6 peak (i.e., 5 to 6 peaks could be baseline separated in an analytical run).

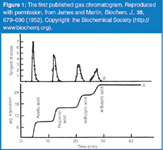
Figure 1
Using GC, chemists could now separate chemical compounds at trace levels, but the challenge was to find an accurate, sensitive and non-discriminating detector. A number of different detectors were developed including the thermal conductivity detector (TCD), coulometric detector and the most important for determination of OC pesticides, the electron capture detector (ECD). The ECD, developed in 1960, was very sensitive towards halogenated compounds and could detect as little as 10–12 moles. It was also very selective for electronegative compounds making it ideal for halogenated organics and organochlorine pesticides.24
Silent Spring
The amounts of pesticides used in the late 1950s and early 1960s was excessive. OC pesticides could significantly increase crop yields by 40% or more. The use of pesticides was seen as positive even though they were highly toxic and persistent. In 1954 and 1956, the reproduction of salmon in the Miramichi River of New Brunswick, Canada, was almost eliminated by the spraying of nearby forests with DDT to control spruce bud worm. In 1962, 350 million pounds of pesticides were used in the US. One of every 12 acres was treated with pesticides. These treatments had a startling affect on wildlife in the US25 and around the world.26 That same year Silent Spring,27 a book by Rachael Carson describing the dangers and effects of pesticides, was published. The title of the book was in response to great numbers of bird deaths as a result of poisoning from their diet of contaminated insects and earthworms. Carson's book indicated that if levels of pesticides continued to increase, birds would eventually disappear and the chirping of birds may not have ever been heard again. By 1973, Dieldrin was detected in 96% of the samples in a food basket study and in 99.5% of humans tested at a lipid adjusted level of 0.3 ppm.28
In the analysis of OC pesticides in the early 1960s, a number of additional peaks were observed in the GC–ECD chromatograms. Figure 2 shows up to 14 unknown peaks that were initially suspected to be OC pesticide degradation products. Jensen analysed archived bird feathers dating back to 1888 and showed the unknown peaks were present prior to 1945 and, therefore, could not be originating from OC pesticides. He used gas chromatography mass spectrometry (GC–MS) to determine that these unknowns were a homologous series of unsaturated organic compounds that contained only carbon, hydrogen and chlorine with molecular weights of 324, 358, 392, 426.29 Figure 3 shows chromatograms for a variety of wildlife from different food web levels. The interfering compounds identified as PCBs were present in every sample. Many early OC results were probably biased due to contribution from PCBs.

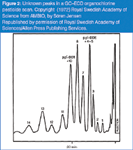
Figure 2
Sample extracts had to be split into fractions to eliminate bias from PCB and OC pesticide overlapping peaks in packed column chromatograms. Extracts were cleaned and fractionated using open column chromatography. Florisil is the most common chromatographic adsorbent, but silica gel and alumina are also used. The majority of the PCBs and a few OCs (typically Aldrin) elute in the first fraction while the remaining OCs and some PCBs (typically the non-ortho substituted PCBs) elute in the second fraction.

Figure 3
Chromatographic and Column Technology
Analyses were performed mainly on packed columns until the early 1980s. Analytical run times were hours long; peaks were up to 10 min wide with many coelutions due to low resolving powers (typically in the order of a thousand). The capillary column, a wall-coated open tubular column, was developed in 1958,30 but due to issues with inertness (metal supports) and fragility (glass supports), it was not used routinely until the invention of fused silica in 1979.31 Fused-silica columns based on optical fibre technology, are inert, flexible and durable.
Stationary phases coat the inner column wall and column lengths can be much greater than packed columns because there are no back pressure issues from particles. Initial column bleed problems were solved by crosslinking stationary phases to the columns. Peak capacities were increased by an order of magnitude from packed columns (to about 50). Capillary columns use much lower gas flows and, therefore, can be directly coupled with mass spectrometers. The separating power of GC and the selectivity of mass spectrometry make GC–MS the universal analytical technique.32,33 Although the mass spectrometer can distinguish between most chemical compounds by mass, it cannot distinguish isomers (which have the same mass) from one another for groups of compounds such as PCBs, PCNs and dioxins. Complete separation of the toxic compounds from the non-toxic ones is a very important consideration when the degree of toxicity varies significantly between isomers and congeners of a group of compounds.
Modern Chromatographic Methods
GC–MS is required to detect dioxins. Dioxins are one of the most toxic chemical groups known to man. They were initially identified as highly toxic by-products of phenoxy-acid herbicides, 3,4,5,-T and 2,4-D, used to control weeds and excess foliage. Dioxins and structurally related compounds, furans, are a group of 210 planar compounds with two chlorine substituted benzene rings connected by one (furan) or two (dioxin) oxygen atoms as shown in Figure 4. Congeners with chlorines in the 2,3,7,8 ring positions (17 congeners) react with the mammalian aryl hydrocarbon receptor (AhR) promoting a number of toxic effects including cancer, immune impairment, as well as developmental and reproductive disorders.34 Congeners without a chlorine in one or more of the 2,3,7,8-positions (193 congeners) are not toxic and do not bioaccumulate in fish and mammals. The LD50 (median lethal dose) for 2,3,7,8-TCDD can be as low as ~1 μg/kg of body weight (guinea pig) while the NOEL (No Observable Effect Level) for 1,3,6,8-TCDD is 3 g/kg body weight. These two compounds are the main dioxin by-products in the formation of the phenoxy acid herbicide: Agent Orange. Compounds that are planar, can be positioned in a rectangle with dimensions of approximately 0.3 ×1.0 nm (10–9 m) with chlorines positioned near the corners of the rectangle, have a higher probability of binding to the AhR and exhibiting dioxin-like characteristics. Some PCBs and PCNs also have significant dioxin-like character.

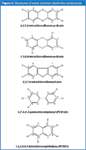
Figure 4
A six-year study (1972–1977) showed that human miscarriages were three times greater in areas where Agent Orange was used35,36 and as such, it was banned in the US in 1979. In 1970 a group of scientists determined that a detection limit of 1 ppt would be required to minimize risk to dioxin exposure. This was much lower than the 50 ppb level achievable at that time and would require a very sensitive and selective analytical procedure with concentration factors of 106–108 to meet detection limits. Gas chromatography high resolution mass spectrometry (GC–HRMS) would potentially be the only technique that could meet these very stringent data quality objectives. The analysis of dioxins is one of the most challenging pursuits in analytical chemistry and a benchmark for the analysis of other organohalogens.37,38 The analytes of interest must be quantitatively extracted from the sample. Interfering compounds must be removed using a series of column chromatographic procedures such as silica, alumina, Florisil or carbon. The extract must then be concentrated to minute volumes to detect sub-picogramme (10–12 g) levels in samples. Interfering compounds including non-toxic isomers and congeners must be separated and the toxic components detected using high resolution mass spectrometry (HRMS).
The use of capillary chromatography with ECD or MS detection has been the standard for the analysis of organohalogens since the early 1980s. Capillary columns are produced in a variety of dimensions. The most common dimensions are 60 m or 30 m columns with 0.25 mm inner diameter (i.d.), 0.25 μm film thickness (f.t.) and non-polar (dimethylsiloxane) or slightly polar (5% diphenlymethylsiloxane) coatings. Most analysts attempt to run all of their analyses on these phases because they provide good separation for the majority of compounds and the phases are inert to the majority of organohalogens and other matrix coextractables. Analytical runs typically are 30–60 minutes in length.
Advancing Chromatographic Methods
Microbore columns (i.d. < 0.18 and f.t. < 0.2) can significantly reduce analysis times. If the phase ratio (the ratio of i.d. to f.t.) is kept constant, the chromatography (relative elution order) and area under the peaks theoretically remain the same. Reducing i.d. and f.t. results in shorter retention times and taller and narrower chromatographic peaks. This technique, called fast GC, can reduce analysis times by 50% or more and increase signal-to-noise (S/N) by up to a factor of five.39–48 The major limitation of this technique is finding a detector that can scan fast enough in order to accurately define a peak (obtain a minimum of 7 to 10 sampling points across a GC peak).49 Figure 5(a) shows the GC time-of-flight mass spectrometric (GC–TOF-MS) separation of 117 compounds — a mixture of PCBs, OCs, chorobenzenes (CBs) and polycyclic aromatic hydrocarbons (PAHs) — achieved in about 7 min. Peaks are about 2 s wide and, therefore, at least 5 scans per second are required to obtain the minimum number of sampling points.

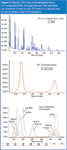
Figure 5
The time-of-flight (TOF) mass spectrometer was the first to be combined with a gas chromatograph.32 Early models had unit mass resolution and were not controlled via a data system. New configurations can scan much faster and have reflectron designs that increase resolution significantly.50
Some data systems now enable deconvolution of overlapping peaks. As shown for the peaks plotted in Figure 5(c), the slope of the tangent of all ions at a specific time can be determined and grouped. Compounds with varied differentials can be deconvoluted from one another. Figure 5(b) shows the total ion chromatogram (TIC) over a 10 s section of a standard containing over 100 OC pesticides, PCBs and PAH. There appears to be five peaks in the Figure 5(b) chromatogram, however, after deconvolution of the peaks is performed, nine peaks can be detected.
Table 2 describes the potential time savings using fast GC. Additional time savings can be realized with analyte-specific columns. Columns for a variety of compounds including dioxins [DB-Dioxin (Agilent Technologies, Santa Clara, California, USA), Rtx-Dioxin2 (Restek, Bellafonte, Pennsylvania, USA)], SP-2331 (Supelco, Bellafonte, Pennsylvania, USA) and BPX-DXN, (SGE, Austin, Texas, USA)]; PCBs [HT-8 (SGE) and Rtx-PCB (Restek)]; and OC Pesticides [Rtx-CLPesticides (Restek)] have been developed.


Table 2: Fast GC time savings.
These columns have been designed to separate critical pairs of coeluting analytes and minimize the number of coelutions seen on dimethylsiloxane or 5% diphenylmethylsiloxane columns. In past years, the adoption of fast GC has been sluggish as older GC ovens could not heat at high enough ramp rates, columns were not readily available in fast GC dimensions (i.d. < 0.18 and f.t. < 0.2) and some regulatory methods were not flexible enough to allow the use of microbore columns. The number and phases of columns available in dimensions capable of doing fast GC has increased significantly in the past few years and newer GCs are all capable of fast oven heating.
Analytical Challenges
Coelution of analytes for the analysis of all groups of halogenated organics presents a significant analytical challenge. There currently is no single GC column that can separate all the components of compound groups such as PCBs, PCNs, dioxins/furans, or OC pesticides in a single analytical run. GC–MS or GC–HRMS is currently the method of choice for dioxins/furans, dioxin-like PCBs and PCNs. OC pesticides and PCBs are also routinely determined using dual column analysis with ECD detection. Multidimensional or comprehensive two-dimensional chromatography (GC×GC) is a relatively new technique that can analyze samples on two different GC phases in the same analysis.51–62 In GC×GC, two different chromatographic columns are connected in series through a modulator, which traps the analytes eluting from the primary column and re-injects them in small compressed packets onto the secondary columns. Figure 6 shows two coeluting peaks eluting from the primary column [6(a)], the resulting modulated packets eluting from the secondary column [6(b)] and the corresponding two-dimensional colour intensity plot or three-dimensional contour plot [6(c)] illustrating the data.


Figure 6
GC×GC has a number of advantages over single column techniques. When orthogonal columns are coupled in series (columns that provide separation through different physical and chemical properties, e.g., boiling point/polarity versus shape selection) separations are ordered in chromatographic space. Thus, different compound groups tend to separate into distinct ordered bands of peaks eluting at about a 45° angle relative to each other. This spatial ordering of compound class peaks aids in the separation, identification and classification of multi-component compound groups (e.g., PCBs, PCNs, dioxins). As indicated earlier, mass spectrometers cannot separate isomers, congeners or homologues with identical mass to charge ratios. GC×GC provides much greater chromatographic resolution and peak capacity than single column systems. The GC×GC peak capacity is the product of the individual peak capacities of the primary and secondary columns and is typically on the order of 1000 peaks or more. With such great separating efficiency, a simple detector such as ECD can be used. Figure 7 shows the GC×GC chromatogram of a number of halogenated organics. GC×GC can also be used as screening method for variety of halogenated compounds.


Figure 7
Polychlorinated alkanes (PCAs) including short chain chlorinated paraffins (SCCPs) are a very challenging group of compounds to analyse.63 They have replaced PCBs in many applications. There are many thousands of congeners and PCAs often appear as a plethora of indistinct peaks creating an unresolved hump in one-dimensional chromatograms (similar to that seen for complex hydrocarbon mixtures). In two-dimensional chromatograms, they appear as structured bands and can easily be identified and quantified. Polychlorinated terphenyls (PCTs) manufactured as Aroclors where used in similar applications as PCBs, but also as synergistic agents for the application of Lindane.64 Not normally detected in conventional analyses, PCTs are often observed in GC×GC chromatograms of sludges and sediments. [See Figure 7(c) and as confirmed using GC×GC–TOF-MS in Figure 8.]

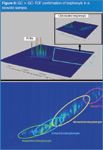
Figure 8
The majority of the halogenated organic compounds listed in Table 1 are lypophilic and have similar physical and chemical properties. They can be extracted together and the extract can be cleaned using common procedures.65,66 Silica, alumina and Florisil are classical procedures used to remove lipids and other polar matrix compounds.67 This results in the ability to collect similar compounds (halogenated POPs) in the same extract and creates the potential for one analytical system to identify and quantify them in a single analytical run. Hyötyläinen68 has reviewed a series of novel sample extraction and chromatographic techniques that can be used to simplify and speed up analyses.
Advancing into the Next Century
Over the last century, sensitivity and selectivity for the determination of halogenated organics has increased significantly. Current analytical methods, in most cases, are selective and sensitive enough to inform scientists of the presence and levels of distinct organohalogens in samples. This helps us in our endeavour to protect human life and wildlife from exposures to toxic persistent organic compounds. The current challenge is to chromatographically separate, detect and quantify as many persistent organic pollutants as possible within the same sample extract. Multidimensional chromatography provides the best chromatographic separation. The mass spectrometer is currently the most selective and sensitive detector possible. Unfortunately, there currently is no mass spectrometer that can scan and detect compounds at the low femtogramme level at rates of 20 s per decade with resolutions of 10000 or better. The ultimate instrument would use GC×GC for separation, with the scan speeds of TOF mass spectrometers (>25 per second) and the selectivity (resolution >10000) and sensitivity (10 fg) of a high resolution magnetic sector instrument. Jensen's 1972 Ambio paper, The PCB Story,29 ends with a compelling moral:
The accumulation of PCB in nature was discovered accidentally, as was mercury contamination. What conclusions can be drawn from these findings? Similar discoveries of the accumulation of other chemicals are quite likely to occur at any time. This should serve to emphasize the need for close cooperation between ecologists, chemists and other scientists. It is necessary that responsible authorities invest in manpower and equipment to facilitate an unbiased search for pollutants at an early stage by systematic analysis.
These are the measures that should be taken if the damaging and perhaps irremediable effects of a substance are to be discovered before and not after it has entered the environment. This applies especially to substances that accumulate and are highly persistent, as we have learnt from the history of PCBs.
Unfortunately, since 1972 there have been numerous troubling discoveries of other chemicals in the environment, in humans and in wildlife. These include brominated compounds68–70 (polybrominated diphenylethers, hexabromocyclododecane), chlorinated compounds71–73 (chlorinated diphenyl ethers, Dechlorane Plus and other Dechlorane compounds) and fluorinated compounds73–75 (PFOS, PFOA).
Regulations such as EU No 850/2004 and the Stockholm Agreement have been developed to reduce or eliminate the use of toxic, persistent and bioaccumulative compounds and reduce levels of these compounds in the environment. Many countries have drafted or are in the process of drafting regulations directed toward the virtual elimination of these compounds in the environment. Even so, the number of chemicals used in commerce and industry increases exponentially every year. Many of these compounds and their degradation products find their way into the environment enabling them to weave their way into every living thing. The ultimate goal would be to develop analytical methods that can analyse all POPs in a single sample extract as well as separate and detect the toxic components at the low femtogramme levels.


Table 3(a): Summary of methods for analysis of halogenated organics (1800sâ1980s).
Over the last century, the advances in analytical methods have been extensive [Table 3(a) and Table 3(b)]. Single toxic components can be isolated from an indefinite number of other chemically similar compunds and accurately quantified at trace levels. Detection limits have decreased by over 10 orders of magnitude. Methods have become more sensitive and selective. Unfortunately, this has also resulted in analysts often being too focused at looking for only their specific compounds of interest. The newer methods: GC×GC and fast scanning detectors should enable us to look at many more compounds in our sample extracts. We must develop these techniques as an analytical triage to assess what hazardous compunds may be present in the sample. There are many thousands of halogenated organic chemicals used by man and many others that are produced as unintentional by-products or degradation products that could potentially be more toxic than the parent compound. Considering how complex the environmental chemistry, fate and transport of these compounds are,75–78 it is vital that we continue to advance our analytical methods to better enable us to scan the environment for persistent toxic organic compounds.
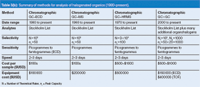
Table 3(b): Summary of methods for analysis of halogenated organics (1980âpresent).
Dr Eric Reiner is a senior mass spectrometry research scientist at the Ontario Ministry of the Environment, Canada. He also serves as an adjunct professor at the University of Toronto and at the University of Waterloo. He has over 20 years experience in the analysis of halogenated persistent organics.
Dr Adrienne Boden is a dioxin and toxic organics scientist at the Ontario Ministry of the Environment, Canada. She performed her PhD studies in chemistry with a focus on environmental analysis at McMaster University. Her current work focuses on the GC×GC analysis of persistent toxic organics.
Tony Chen is a senior toxics organic scientist at the Ontario Ministry of the Environment with 20 years experience in the analysis of OC pesticides, PCBs and PAH.
Karen MacPherson is a senior dioxin scientist at the Ontario Ministry of the Environment with more than 20 years of experience in the analysis of Dioxin-like compounds and brominated flame retardants.
Alina Muscalu, MASc, is a toxic organics technologist at the Ontario Ministry of Environment. She has 7 years experience in the analysis of PCBs, OC Pesticides. She has been working with GC×GC for over 2 years and is currently completing her PhD at the University of Waterloo.
References
1. Stockholm Convention Secretariat, 2001, UNEP, http://chm.pops.int/
2. D.G.C. Muir and P.H. Howard, Environ. Sci. Technol., 40, 7157–7166 (2006).
3. R. Lohman et al., Environ. Pollut., 150, 150–165, (2007).
4. R. Bethem et al., J. Am. Soc. Mass Spectrom., 14, 528–541, (2003)
5. J.S. Waid, PCBs in the Environment, Vol I, II and III (CRC Press, Boca Raton, USA, 1986).
6. D. Hayward, Environ. Res., 76, 1–18, (1998).
7. Organic Analytical Chemistry: Theory and Practice: Mohan, G, Alpha Science International, Pangbourne, England, 2003, pg 529.
8. W.A.Cook and K.H. Cook, Ind. Eng. Chem., 5, 186–188, (1933).
9. C.W. Bacon, J. Am. Chem. Soc., 31, 49–52, (1909).
10. K. Herxhiemer, Muench. Med. Wochenschr., 46, 268 (1899).
11. C.K. Drinker, M.F. Warren and G.A. Bennet, J. Ind. Hygiene Toxicol., 19, 283–311 (1937).
12. W.B. Crummett, Decades of Dioxin: limelight on a molecule, (Xlibris, Philadephia, USA. 2002).
13. R. Carson, Silent Spring, (Houghton Miffin, Boston USA. 1962) Chpt. 3
14. M. Gladwell, The New Yorker, July 2, 42–51 (2001).
15. V. Zitko, Chlorinated Pesticides: Aldrin, DDT, Endrin, Dieldrin, Mirex, H. Fiedler, ed, (Springer Verlag, Berlin, 2003) 47–90.
16. M.S. Schechter et al., Ind. Eng Chem., 17, 704–709 (1945).
17. M. Tswett, Ber. Dtsch. Botan. Ges., 24, 316–323 (1906).
18. D.T, Day, Science, 17, 1007–1008, (1903).
19. J.V. Hinshaw, LCGC North America, 21, 546-549 (2003).
20. A.P.J. Martin and R.L.M. Synge, Biochem. J., 25, 1358-1368 (1941).
21. A.T. James and A.J.P. Martin, Biochem. J., 35, 679-690 (1952).
22. V.J. Cirillo, J. Chromatogr., 81, 197-205 (1973).
23. K.D. Bartle and P Myers, Trends Anal. Chem., 21, 547–557 (2002).
24. J.E. Lovelock, Anal. Chem., 33, 162–178 (1961).
25. D.S. Greenberg, Nature, 140, 878–879 (1963).
26. C. Bernes, Persistent Organic Pollutants: A Swedish View of an International Problem, (Swedish Environmental Protection Agency, Stockholm, 1998)
27. R. Carson, Silent Spring, (Houghton Miffin, Boston USA. 1962).
28. Time, Environment: The Dieldrin Dilemma, May 2, 1974.
29. S. Jensen, Ambio, 1, 123–131 (1972).
30. D.H. Desty, H.H. Naresnape, B.H.F. Whyman, Anal. Chem., 32, 302-304 (1960).
31. K. Grob, Chromatographia, 8, 423–433 (1975).
32. R.S. Gohlke, Anal.Chem., 31, 535–541 (1959).
33. J.T. Watson and K Biemann, Anal. Chem., 37, 844–851 (1965).
34. A.B. Okey, Toxicol. Sci., 98, 5–38 (2007).
35. C. Cookson, Nature, 278, 108 (1979).
36. A. Hay, Nature, 278, 108–109 (1979).
37. E.J. Reiner et al., Anal. Bioanal. Chem., 386, 791–806 (2006).
38. E.J. Reiner, Mass Spectrom. Rev. DOI 10.1002/mas.2055 (2010).
39. M. Domotorova and E. Matisova, J. Chromatogr. A., 1207, 1–16 (2008).
40. P. Korytar et al., Trends Anal. Chem., 21, 558–572 (2002).
41. C.A. Cramers and P.A. Leclercq, J. Chromatogr. A., 842, 3–13 (1999).
42. K. Mastovska and S.J. Lehotay, J Chormatogr. A., 1000, 153–180 (2003).
43. M.S. Klee and L.M. Blumberg, J.Chromatogr. Sci., 47, 83–91 (2002).
44. E. Reiner et al., Organohaogen Compd., 66, 825-832 (2004).
45. P. Sandra and F. David, J.Chromatogr. Sci., 40, 248–253 (2002).
46. J. Cochran, Chromatogr. Sci., 40, 254–268 (2002).
47. E.J. Reiner et al., Organohaogen Compd., 45, 17-20 (2000).
48. K.A. MacPherson et al.,Organohalogen Compd., 40, 19–23 (1999).
49. P.A. Leclercq and C.A. Cramers, Mass Spectrom. Rev., 17, 37–49 (1998).
50. N. Mirselah-Kohan, W.D. Robertson and R.N. Compton, Mass Spectrom. Rev., 27, 237–286 (2008).
51. Z. Liu and J.B. Phillips, J. Chromatogr. Sci., 29, 227–231 (1991).
52. J.B. Phillips and J. Xu, J. Chromatogr. A., 703, 327–334 (1995).
53. M. Adahchour et al., TrAC, Tends Anal. Chem., 25, 726–741 (2006).
54. M. Adahchour et al., J. Chromatogr. A., 1186, 67–108 (2008).
55. L.R. Bordajandi et al., J. Chromatogr. A, 1186, 312–324 (2008).
56. C. Danielsson et al., J.Chromatogr. A., 1086, 61–70 (2005).
57. J.F. Focant et al., Talanta, 63, 1231–1240 (2004).
58. J.F. Focant et al., Anal. Chem., 76, 6313–6320 (2004).
59. P. Haglund et al., Anal. Bioannal. Chem., 390, 1815–1827 (2008).
60. L. Mondello et al., Mass Spectrom. Rev., 27, 101–124 (2008).
61. O. Panic and T. Gorecki Anal. Bioannal. Chem., 386, 1013–1023 (2006).
62. J.V. Hinshaw, LCGC North America, 22, 32–40 (2004).
63. E. Eljarrat and D. Barcelo, TrAC. Anal. Chem., 25, 421–434 (2006).
64. E.J. Duda, J. Econ. Entom., 50, 218–219 (1957).
65. J. Boer, J. Chromatogr. A., 843, 179–198 (1999).
66. S.P.J. Van Leeuwen and J. de Boer, J. Chrom A., 1186, 116–182 (2008).
67. S.K. Poole et al., Anal. Chim. Acta, 236, 3–42 (1990).
68. T. Hyotylainen, LCGC Europe, 22, 173–179 (2009).
69. T.M. Kolic et al., J. Chromatogr. Sci., 47, 83-91 (2009)
70. C. de Witt, Chemosphere, 46, 583–624 (2002).
71. M. Becker, T. Phillips and S. Safe, Toxicol Environ Chem., 33, 189–200 (1991).
72. E. Hoh, L. Zhu and R.A. Hites, Environ. Sci. Technol., 40, 1184–1189 (2006).
73. L. Shen et al., Environ. Sci. Technol., 44, 760–766 (2010).
74. R. Renner, Environ. Sci. Technol., 35, 154A–160A.
75. J.P. Giesy and K. Kannan, Environ. Sci. Technol., 36, 146A–152A (2002).
76. F. Wania, Environ. Sci. Pollut. Res., 6, 11–19 (1999).
77. T. Meyer, F. Wania and K. Breivik, Environ. Sci. Technol., 39, 3186–3196 (2005).
78. K. Fenner et al., Environ. Sci. and Technol., 39, 1932–1942 (2005).
Note: Organohalogen papers can be downloaded from: www.dioxin20XX.org















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.

Miniaturized GC–MS Method for BVOC Analysis of Spanish Trees
April 16th 2025University of Valladolid scientists used a miniaturized method for analyzing biogenic volatile organic compounds (BVOCs) emitted by tree species, using headspace solid-phase microextraction coupled with gas chromatography and quadrupole time-of-flight mass spectrometry (HS-SPME-GC–QTOF-MS) has been developed.

