A Comparative Study of Various Sample Preparation Approaches for PFAS in Biofluids: A Practical Method of Biomonitoring for Anthropogenic Toxicity
Per- and polyfluoroalkyl substances (PFAS) constitute a diverse class of highly persistent anthropogenic chemicals detectable in the biofluids of most individuals. Known as “forever chemicals”, PFAS exposure has been linked indirectly and directly to a range of potential health issues. Ongoing research aims to uncover the probable link between PFAS concentrations in the body to a wide range of adverse health outcomes. The development of a robust biomonitoring method for an expanded 34 PFAS panel, closely aligned to other environmental monitoring regulations, is therefore highly advantageous for clinical research because it would allow for accurate measurement of PFAS. The objective of the current study was the development of an optimized sample preparation and liquid chromatography–tandem mass spectrometry (LC–MS/MS) method for the analysis of PFAS in serum for biomonitoring applications.
Multiple studies have recently reported an association between per- and polyfluoroalkyl substances (PFAS) exposure and adverse health outcomes. This has triggered a growth in activities focused on understanding and measuring PFAS exposure levels and the probable impact of PFAS toxicity on the human body. There has therefore emerged an acute need for biomonitoring of PFAS (1). The current CDC procedure measures 17 PFAS analytes in serum (2,3). However, over time, adverse health effects have been identified in relation to more PFAS than originally studied, as a result of exposure through various sources such as food grown in contaminated environments, packaging, cooking pans, and drinking water. Consequently, the expanded panel of 34 PFAS reported in this study aims to provide a more comprehensive assessment, aligned with standardized methods for testing drinking water for these persistent chemicals (see Table 1). The objective was to provide a method for establishing a direct correlation between bioaccumulation of the “forever chemicals” possibly ingested through potable water and the potential health impacts. These chemicals undergo a very slow breakdown process and are known to build up in people, animals, and the environment over time. PFAS can be measured in multiple biological matrices, including but not limited to whole blood, serum, plasma, urine, breast milk, tissues, and hair. Quantitation of a large panel of PFAS is challenging because of the varied chemical nature of the molecules. The preferred analytical technique to efficiently detect and quantitate trace levels of PFAS for biomonitoring is liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS).



Efficient LC–MS/MS for PFAS analysis requires optimized sample preparation to ensure quantitative recovery of the analytes. There are reports of using ion-pair liquid-liquid extraction (LLE) and protein precipitation followed by graphitized carbon black (GCB) cleanup as a sample preparation for extractable organic fluoride in serum (1), while in recent years, environmental methods for the estimation of PFAS have shown success employing solid-phase extraction (SPE) using mixed-mode weak anion exchange. This method has been extended to extracting PFAS from biological samples as a standalone process or in conjunction with other techniques, resulting in limited success over a smaller range of PFAS. The reason for this difference in outcome between PFAS extraction in drinking water and in biofluids may be attributed to the difference in complexity in the sample matrices. The drawbacks to the methods described above include challenges in recovering all PFAS quantitatively from retentive ion-exchange SPE media, and that PFAS bind to GCB during dispersive SPE. The approach detailed here was aimed at designing a practical method for accurate estimation of PFAS in biological samples consisting of an effective sample preparation method that works for a diverse PFAS panel followed by a robust LC–MS method for accurate and reproducible quantitation. The extraction efficiencies of retentive and non-retentive sample preparation techniques for 34 PFAS were compared.
Experimental
An Expedient Non-Retentive Sample Preparation Method
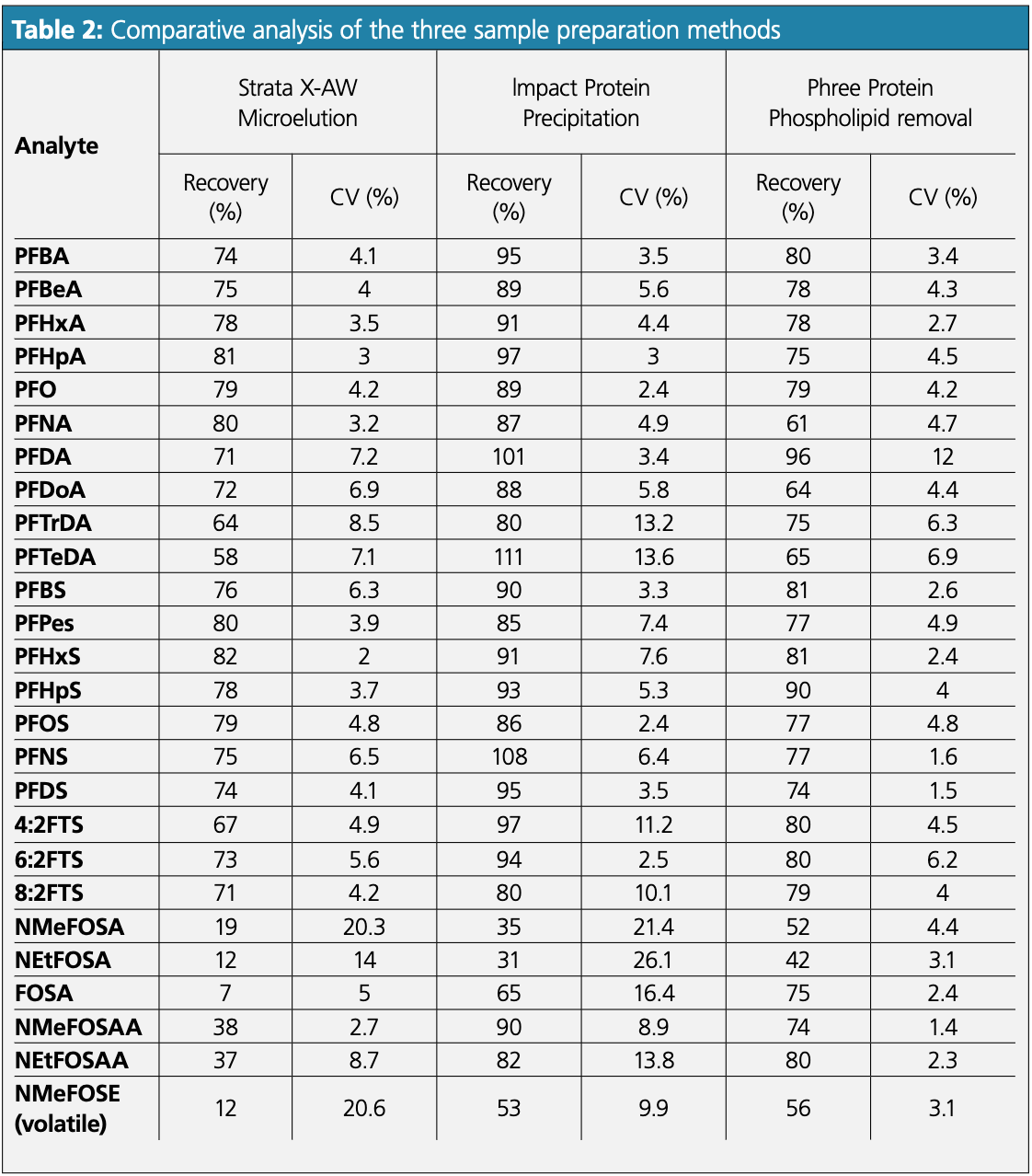
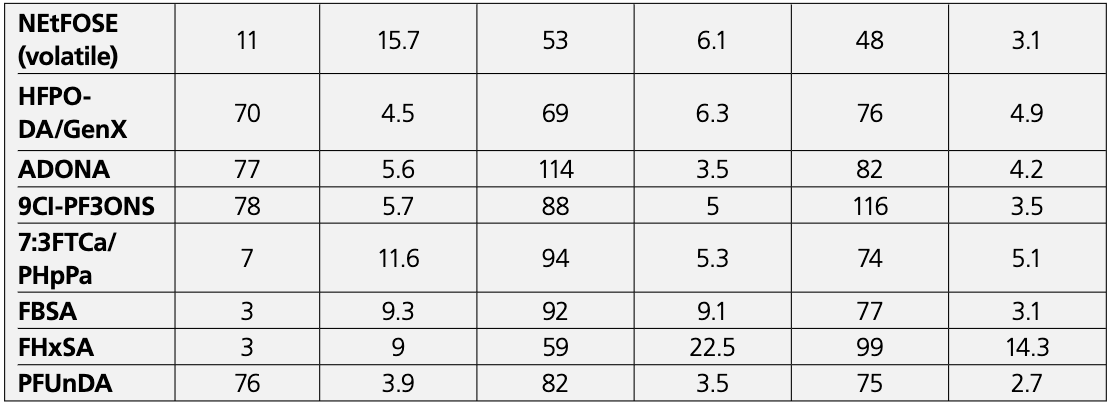
A non-retentive sample preparation method was optimized using Bovine serum albumin (BSA) and a commercially available human serum with the lowest PFAS background available (Mass Spect Gold cat# 3200, from GoldenWest Diagnostics LLC). Serum aliquots were spiked with a standard PFAS mix to compare the recovery using weak anion exchange (WAX) SPE, protein precipitation, and one-step in well protein precipitation and phospholipid removal methods. Average recoveries were calculated from four replicates (N = 4) for each analyte.
Optimized Sample Preparation Method
A 100 μL measure of human serum was added to 500 μL of 1% formic acid-acetonitrile mixture at 0 0 C in a Phree 96 well plate (Phenomenex) (PFAS spike concentration used: 10 ng/mL). The mixture was vortexed for 15 s. After allowing 5 min for completion of protein precipitation, the sample was passed through the plate and collected under vacuum. It was then diluted with 500 μL of 5 mM ammonium acetate spiked with 20 μL of 125 ng/mL internal standard. %CV for all sample preparation experiments used four replicates (N = 4).
Sample preparation experiments were performed using Phenomenex 96 well plates: Phree 96 well plates: 8E-S133-TGB, Strata X-AW Micro-elution plates: 8M-S038-4GA.
Optimized LC–MS/MS Method for Analyte Identification
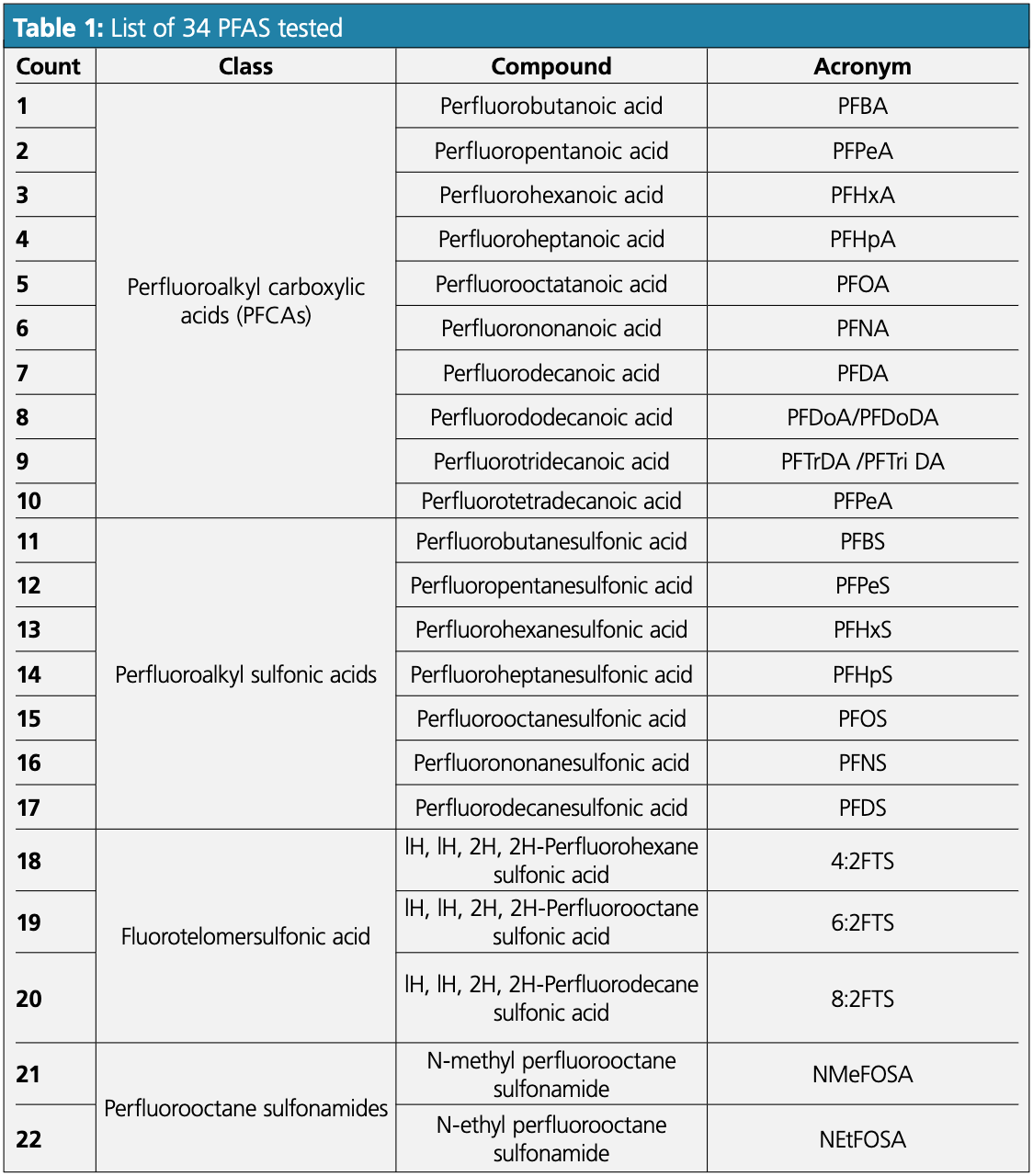
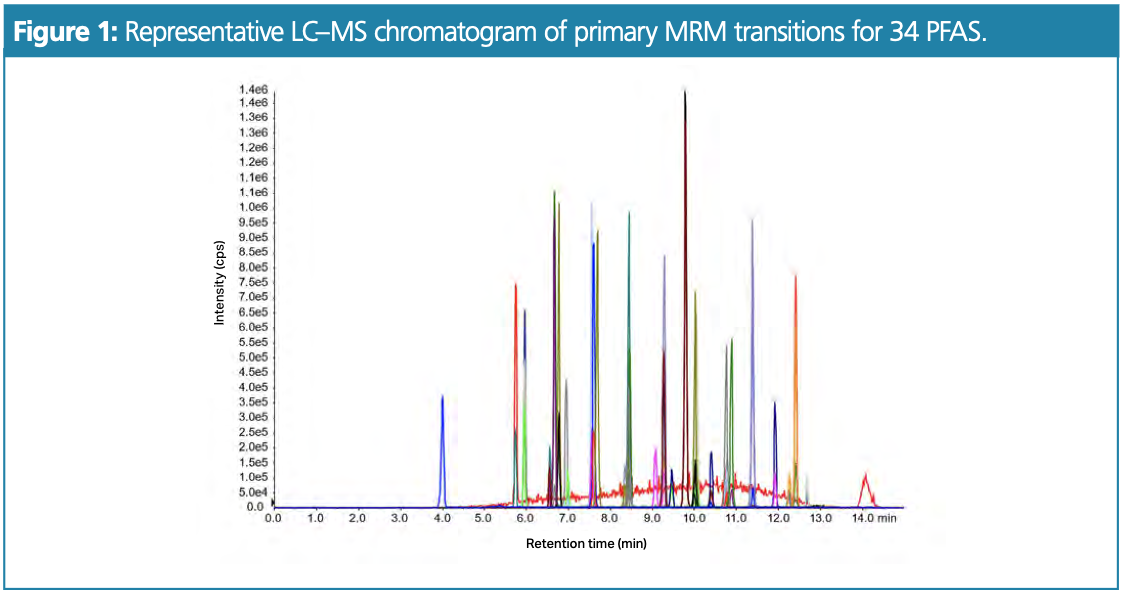
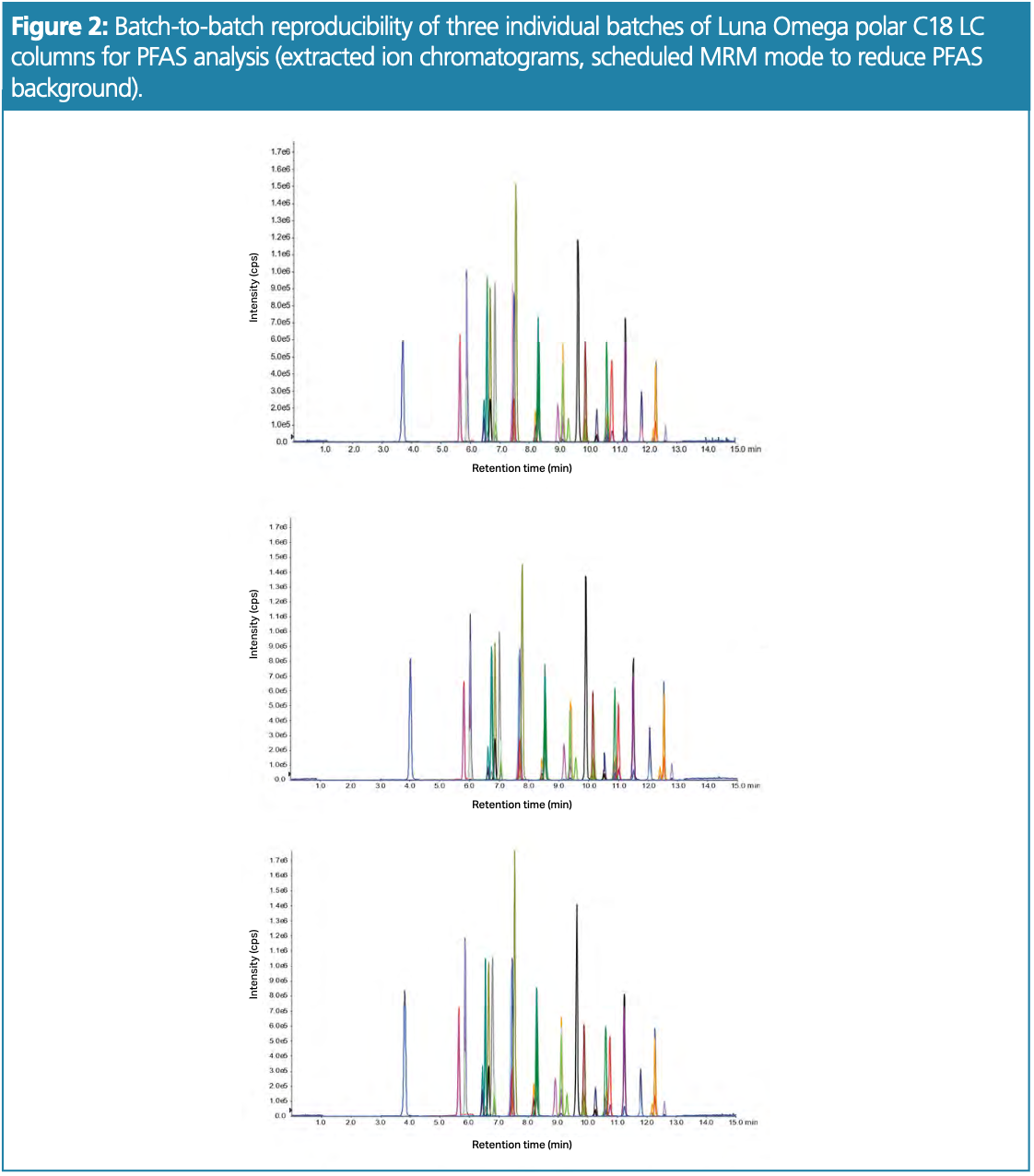
Three reverse-phase 50 × 3.0 mm, 3.0-μm Phenomenex Luna Omega Polar C18 100 Å LC columns from individually manufactured batches were evaluated using the optimized LC–MS/MS method (see Figures 1 and 2) to ensure reproducibility and robustness.


The LC–MS/MS system consisted of an Agilent 1200 liquid chromatograph coupled with a AB Sciex Q-Trap 5500 mass spectrometer equipped with a negative polarity electrospray ionization (ESI) source. To reduce background and delay PFAS contamination from mobile phases, an Agilent PFC-free LC conversion kit was installed.
The following conditions were used: mobile phase: A: 5 mM ammonium acetate, B: methanol; gradient: 0.0 min 5%B, 4.0 min 55%B, 11.0 min 85%B, 11.1 min 95%B, 12.0 min 95%B, 12.5 min 5%B, 15.0 min 5%B; flow rate: 500 μL/min; analyte conc.: 5 ng/mL; injection vol.: 5 μL; temp.: 40 °C.
Results and Discussion
A thorough comparative study was performed using various sample preparation methods that are commonly employed for PFAS extraction (see Table 2). Average percent recoveries from protein precipitation were found to be superior to the WAX SPE method, demonstrating that a simple protein precipitation is a better choice for the more diverse panel of the 34 PFAS. This could potentially be the result of differences in chemical interaction with the SPE media and retention patterns. Improved recoveries and precision (%CV) for all analytes were achieved with a method refinement using a single step in well protein precipitation combined with phospholipid removal. The advantage of this approach is that it requires minimal method development and is easy to implement in most laboratory workflows. However, it should be noted that intrinsic PFAS contamination remains a significant challenge (4,5). It was observed that commercially available sera from the same source but different lots showed low concentrations of multiple PFAS, potentially because of contamination during collection and processing. It is expected in the future that serum samples with lower background concentrations of PFAS will be available, and methods will be optimized further to achieve more accurate quantitation at low concentrations.


The LC–MS/MS conditions reported above can be utilized for expanded panels of PFAS, both diverse in their chemical nature and molecular structures.
Conclusion
A comparative study of different sample preparation approaches for an expanded panel of 34 PFAS demonstrated that a single step in well protein precipitation with phospholipid removal is a simple and elegant sample preparation protocol requiring minimal method development. This method provided significantly improved overall recovery and precision for 34 PFAS. The current sample preparation method paired with the demonstrated LC–MS/MS method is highly reproducible and can be extended to an automated high-throughput workflow with minimal analytical development effort for biomonitoring. Biomonitoring is currently a topic of interest because of the known and far-reaching toxicological impact of these compounds. The current method was developed using available reagents with variable PFAS background. Wider commercial availability of human serum with minimal PFAS background will improve method optimization and performance.
References
(1) Fisher, F. C.; Ludtke, S.; Thackray, C.; et al. Binding of Per- and Polyfluoroalkyl Substances (PFAS) to Serum Proteins: Implications for Toxicokinetics in Humans. Environ. Sci. Technol. 2024, 58 (2), 1055–1063. DOI: 10.1021/acs.est.3c07415
(2) National Academies of Sciences, Engineering, and Medicine. Guidance on PFAS Exposure, Testing, and Clinical Follow-Up. The National Academies Press, 2022. DOI: 10.17226/26156
(3) CDC Environmental Health, Perfluoroalkyl and Polyfluoroalkyl Substances NHANES 2017-2018.
(4) Kaiser, A.-M.; Aro, R.; Kärrman, A.; et al. Comparison of Extraction Methods for Per- and Polyfluoroalkyl Substances (PFAS) in Human Serum and Placenta Samples—Insights into Extractable Organic Fluorine (EOF). Anal. Bioanal. Chem. 2021, 413, 865–876. DOI: 10.1007/s00216-020-03041-5
(5) Frigerio, G.; Cafagna, S.; Polledri, E.; Mercadante, R.; Fustinoni, S. Development and Validation of an LC– MS/MS Method for the Quantitation of 30 Legacy and Emerging Per and Polyfluoroalkyl Substances (PFASs) in Human Plasma, Including HFPO-DA, DONA, and cC6O4. Anal. Bioanal. Chem. 2022, 414, 1–20. DOI: 10.1007/s00216-021-03762-1
About the Authors
Rajashree Chakravati serves as the Global Product Manager for Phenomenex. Direct correspondence to: rajashreec@phenomenex.com
Stephanie J. Marin is the Clinical and Forensics Global Market Development Manager at Phenomenex.
Shahana W. Huq is a senior application scientists at the Phenomenex Applications Laboratory.









 | Image Credit: © Infinity Lens - stock.adobe.com")
 | Image Credit: © MP Studio - stock.adobe.com")








Characterizing Plant Polysaccharides Using Size-Exclusion Chromatography
April 4th 2025With green chemistry becoming more standardized, Leena Pitkänen of Aalto University analyzed how useful size-exclusion chromatography (SEC) and asymmetric flow field-flow fractionation (AF4) could be in characterizing plant polysaccharides.






