Using Cyclodextrins to Achieve Chiral and Non-chiral Separations in Capillary Electrophoresis
LCGC Europe
Describing the theoretical background on how the separations occur, the operating approaches that can be taken and method development.
What Are Cyclodextrins (CDs)?
Cyclodextrins (CDs) are cyclic oligosaccharides composed of D-glucose units that are linked by α(1,4)-glucosidic bonds. The practically important, industrially produced CDs are α-CD, β-CD and γ-CD which differ in the number of glucose units involved [i.e., α-CD contains 6 glucose units, β-CD has 7 glucose units (Figure 1) and γ-CD has 8 glucose units]. CDs are generally produced through enzymatic conversion of starch. CDs have the shape of a torus with a hydrophobic interior cavity and a hydrophilic outside. The narrow rim is occupied by the primary hydroxyl groups at C6 while the wider rim contains the secondary hydroxyl groups at C2 and C3. The hydroxyl groups can be chemically modified resulting in a number of derivatives. With regard to their use as chiral selectors in CE the most important difference is the cavity size which is the smallest for α-CD and the largest for γ-CD.


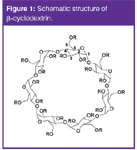
Figure 1: Schematic structure of β-cyclodextrin.
What are CDs used for in CE?
One of the premier applications of capillary electrophoresis (CE) is the resolution of stereoisomers (enantiomers). Enantioseparations by CE are achieved by the addition of a stereochemically pure substance, a so called chiral selector, into the background electrolyte. In order to achieve a chiral separation the enantiomers must have differing chromatographic interactions with the added chiral selector. The most frequently additives used are cyclodextrins because they are commercially available, UV-transparent and relatively low cost.
There is a large variety of CD derivatives that allow optimization during method development as well as the separation of charged and neutral analytes. Many validated enantioseparation methods have been reported. Chiral CE methods for assessing chiral purity and stability are routinely used in many pharmaceutical companies and have also been included in numerous regulatory submissions. Chiral CE methods have also been included in Pharmacopoeias. For example, there is a chiral test method for epinephrine bitartrate in the USP.
Cyclodextrins are also useful in non-chiral separations. For example, they can be used to separate closely related compounds such as cis- and trans-isomers, which have virtually identical electrophoretic mobilities. These compounds will, however, have differing interactions with the CD's because of their differing shape. For example, a range of closely related impurities of the drug salbutamol were resolved from each other and from the salbutamol enantiomers1 using a low pH buffer containing 100 mM dimethyl-β-cyclodextrin. A more elaborate buffer of phosphate pH 3.0 containing 12 mM tetrapropyl and 8 mM tetrabutyl ammonium bromide and 20 mM methyl-β-CD was required for the separation of very closely-related Remoxipride achiral impurities from each other,2 a separation will be observed in most cases.
How Does Adding CDs Help Achieve Chiral Separation?
As mentioned before CDs can form complexes with molecules based on their inclusion into the hydrophobic cavity. Secondary interactions may include hydrogen bonding or dipole–dipole interactions with the hydroxyl groups on the CDs, or with other polar substituents of the CDs. In the case of charged CDs ionic interactions will also contribute, or may even dominate, the complexation mechanism. Effectively the CDs are contained in the buffer and their interaction with the analyte either slows or increases movement of the enantiomers. If the interaction is stronger for one enantiomer than the other then a separation will occur.
CDs are naturally chiral molecules, therefore, binding of host enantiomer molecules results in diastereomeric complexes. These complexes differ in properties such as the enantiospecific binding constants, charge or size resulting in an enantioseparation. The thermodynamic complexation equilibria between the enantiomers R and S and the CD are characterized by the complexation constants KR and KS, respectively, assuming the formation of a 1:1 complex between the enantiomers and the chiral selector.


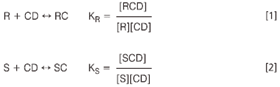
Considering that the effective mobility μeff of an analyte is the sum of its fraction migrating in the free form, μf, and the complexed form, μcplx, and that the effective mobilities of the enantiomers must be different to observe a separation, the fundamental equation as developed by Wren and Rowe can be obtained.3

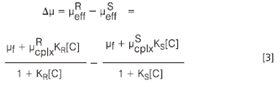
μReff and μSeff are the effective mobilities of the R- and S-enantiomers, μRcplx and μScplx are the mobilities of the complexed R- and S-enantiomers and [C] is the concentration of the CD. As can be derived from Equation 3 enantioseparations can be achieved if the analyte enantiomers differ in their complexation constants or in the complex mobilities, the former is certainly the more often the more dominant factor. Another important conclusion is the fact that the CD concentration plays an important role in enantioseparations.
Cyclodextrins are also used in HPLC to achieve chiral separations where they are bonded onto stationary phase (e.g., Cyclobond columns).
What CDs are Available?
Commercially available CDs are based on the three CDs differing in cavity size (i.e., α-CD, β-CD and γ-CD). The derivatives are obtained by chemical modification of the primary and secondary hydroxyl groups on the CD rims. Most derivatives are based on β-CD but more and more derivatives of α- and γ-CD became available recently. Some commercially available neutral as well as charged CDs are summarized in Table 1. From a practical point of view it is important to keep in mind that many of the CD derivatives are randomly substituted CDs that are essentially a mixture of isomers as this may affect method robustness. Chemically pure single isomer CDs are also available such as the single isomer sulphated CDs developed by Vigh and coworkers in the late 1990s.2–4


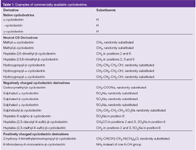
Table 1: Examples of commercially available cyclodextrins.
Who Sells CDs?
The native CDs and several of their derivatives can be obtained from suppliers for organic chemicals such as Sigma, Aldrich or Fluka. A large variety of derivatives can be obtained from CTD Inc. (High Springs, Florida, USA), probably the most complete selection including variations in the degree of substitution and isomeric purity is supplied by Cyclolab (Budapest, Hungaria). Specific CDs are sold by specialized companies such as PAC L.P. (Houston, Texas, USA) providing single isomer sulphated β-CDs or CyDex Inc. (Lenexa, Kansas, USA) commercializing sulphobutylether-β-CD.


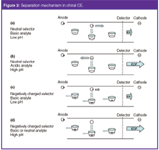
Figure 2: Separation mechanism in chiral CE.
What Separation Modes Can be Applied?
In CE enantioseparations CDs act as pseudostationary chromatographic phase. Thus, depending on the charge of the analytes and the CDs different separation schemes can be applied. The most frequently used approaches are summarized in Figure 2 and in Table 2. In the case of neutral CDs only charged selectors can be separated.



Table 2: Commonly used modes of operation for chiral CE.
When analysing basic compounds acidic background electrolytes are selected [Figure 2(a)]. Under these conditions the analyte is protonated and migrates to the detector at the cathodic end of the capillary while the selector does not possess an electrophoretic mobility but is transported by the EOF. At low acidic pH values the EOF is largely suppressed. Subsequently, the analyte enantiomer, which is complexed stronger by the selector, migrates second as it is complexed for a longer period of time compared with the weaker bound enantiomer. As the hydrodynamic radius of the enantiomer-CD complex is larger than the radius of the free analyte the complex migrates slower. The CD effectively acts as a "net" trapping one enantiomer more than the other and slowing its migration down causing separation to occur.
Acidic analytes can be analysed at alkaline pH values [Figure 2(b)]. Because of their negative charge acids migrate to the anode but are transported to the cathode by the strong EOF at alkaline pH values. In this case the stronger complexed enantiomer migrates first as its mobility in the opposite direction of the detector is slowed down.
Charged chiral selectors are more versatile and offer additional possibilities as they possess an electrophoretic mobility themselves and, as a consequence, neutral molecules can be analysed as well. The possible scenarios are outlined for negatively charged selectors but analogous schemes can be drawn for positively charged selectors. At acidic pH values the negatively charged selector migrates to the anode while positively charged basic analytes migrate towards the cathode where they are detected [Figure 2(c)]. In this instance the stronger complexed enantiomer migrates second as described above for the use of neutral selectors. A general advantage of selectors with the opposite charge compared with the analytes is their counter mobility, which enables the use of low concentrations of the respective chiral selector. If high CD concentrations are selected the analyte may be carried to the anode. In this case the polarity of the applied voltage is reversed and detection is performed at the anode. This will lead to a reversal of the enantiomer migration order.
At alkaline pH values negatively charged CDs may also be applied to the enantioresolution of neutral and basic analytes in the normal polarity mode [Figure 2(d)]. At alkaline pH values basic compounds are uncharged and are transported to the detector at the cathode as are neutral compounds. The anionic CD migrates to the anode and decelerates the stronger complexed enantiomer compared to the weaker complexed enantiomer. Thus, the weaker bound enantiomer is detected first.
Are There Screening Approaches I Can Use in Method Development?
In practice it is common to use a variety of potential operating conditions to assess which operating conditions may prove useful. Standard sets of buffers and CD's may be assessed in an overnight sequence to discover which conditions provide some enantioseparation. These conditions are then optimized in terms of CD concentration and other factors such as buffer concentration/composition. Deeb et al.7 recently used a screen of 22 different CDs to optimize the separation of 14 different chiral compounds. They also systematically evaluated a series of operating parameters such as CD concentration, buffer type/concentration and the addition of organic solvents.
A strategic approach to the development of capillary electrophoresis chiral methods for pharmaceutical basic compounds using sulphated cyclodextrins has also been published.8 More general method development strategies for the enantioseparation of drugs by capillary electrophoresis using cyclodextrins as chiral additives have also been published.9 Generic capillary electrophoresis conditions for chiral assays are often used in early pharmaceutical development and conditions have been published.10,11
Do I Need Highly Pure Cyclodextrins?
Especially pure CDs are not necessarily required for successful enantioseparations. In fact, many reported methods have been achieved using randomly substituted derivatives. However, randomly substituted CDs are a mixture of isomers differing in their degree of substitution (i.e., the number and the position of the substituents). Therefore, randomly substituted CDs from various suppliers may differ in this respect and differences may even be observed from batch-to-batch. Literature examples clearly demonstrate that the source of the CD and the degree of substitution affects the resolution in CE enantioseparations of several compounds while this may have no effect for other analytes. Even reversal of the enantiomer migration order has been observed when switching from a CD with low isomeric purity to a quality with high isomeric purity.12
An illustrative example of the effect of CDs from different sources on the enantioseparation of ropivacaine is shown in Figure 4. While the separation of the minor (R)-enantiomer could be achieved with 2,6-dimethyl-β-CD from 2 suppliers, the product from Supplier 3 failed to resolve the enantiomers under the experimental conditions applied. This is because the CDs differ in their degree of substitution, as not all 2 and 6 positions of β-CD may be derivatives or methylation of the 3-hydroxy groups may have occurred as well. The example clearly demonstrates that variations in the degree of substitution of randomly substituted CDs can affect the robustness of the method and has to be considered during method validation. CDs of various suppliers and different batches should be evaluated during the process. Generally, it cannot be predicted if a higher or lower degree of substitution of a given CD results in a better enantioseparation, examples for both scenarios have been reported. With regard to method robustness single isomer CDs have a clear advantage when equally good enantioseparations are observed for both types of CDs.



Figure 4: Dependence of the enantioseparation of (R)- and (S)-ropivacaine on the source of the cyclodextrin. Experimental conditions: 72 cm, 50 μm i.d. fused-silica capillary, mM sodium phosphate buffer, pH 3.0, containing 13.3 mg/mL 2,6-dimethyl-β-CD, 30 °C, applied voltage 27 kV. (Reprinted with permission from C. Sänger-van de Griend, Enantiomeric Separations by Capillary Electrophoresis in Pharmaceutical Analysis, PhD thesis, Acta Universitatis Upsaliensis, Uppsala 1999, ISBN 91-554-4561-6)
How Much CD Do I Need to Add to Achieve My Separation?
There is no general rule how much of a certain CD is necessary to achieve an enantioseparation as this will vary with the type of the CD, the analyte and the separation problem. The separation depends on the formation of the CD-analyte complexes as well as their electrophoretic mobilities (see Equation 3). Thus, if large differences of the complexation constants between the CD and the analyte enantiomers exist, relatively low concentrations of the CD will suffice to achieve an enantioseparation while higher concentrations may be necessary in the case of small differences between the complexation constants. The strong electrostatic interactions between oppositely charged analytes and CDs often allow the use of very low selector concentrations. For example, in the case of enantioseparations of protonated amines by negatively charged CDs efficient separations at CD concentrations in the range of 0.5–1 mg/mL have been reported.
Sulphated cyclodextrins have multiple negative charges and, therefore, genearate high operating currents if used in high concentrations. Therefore, their use is limited to low concentrations or using narrow bore capillaries and/or cooling devices to limit the current.
If only a chiral separation is desired (i.e., resolution RS = 1.5) lower concentration may be sufficient compared with the determination of the enantiomeric excess when a low concentration of an enantiomeric impurity has to be determined in the presence of a large excess of the eutomer (desired enantiomer). Then higher concentration of CD may be necessary. However, one should keep in mind that optimum CD concentrations may exist resulting in lower resolution after the maximum. The choice of the CD and the optimization of the CD concentrations are among the most important variables in chiral CE.7
What Other Additives Can I Use if CDs Don't Work?
Other chiral selectors are available but have been less frequently used as a result of limited availability, applicability with regard to analytes, or experimental problems including adsorption to the capillary wall. Consequently, only a few validated and robust methods have been reported using these selectors. Macrocyclic glycopeptide antibiotics such as vancomycin or teicoplanin have been successfully applied to the enantioresolution of acidic compounds.13 The chiral crown ether, (+)-(18-crown-6)-2,3,11,12-tetracarboxylic acid, is suitable for enantioseparations of primary amines and amino acids.14 The enantioresolution of amino acids and their derivatives has been achieved by ligand exchange employing copper(II)-complexes of amino acids or amino acid derivatives.15 Chiral ion-pairing reagents such as cinchona alkaloid derivatives or camphorsulphonic acid proved to be effective selectors in non-aqueous capillary electrophoresis.16 Further selectors include linear polysaccharides and proteins. In micellar electrokinetic chromatography (MEKC) and microemulsion electrokinetic chromatography (MEEKC) enantioseparations using chiral detergents such as bile acids or amino acid derivatives have been described.17–19
What Applications Are There?
Enantioselective CE methods have been applied to analytical processes in the chemical, pharmaceutical, food and cosmetics industry as well as in forensic and environmental sciences. A specifically challenging area in pharmaceutical analysis is the determination of the stereochemical purity of a compound when the stereochemical impurity has to be quantified in the presence of a large excess of the other stereoisomer. Overloading the CE system creates problems ranging from peak distortion to loss of resolutions. Nevertheless, chiral CE methods have been successfully developed and validated.


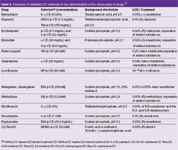
Table 3: Examples of validated CE methods for the determination of the chiral purity of drugs.12
Some recent examples are summarized in Table 3. It can be clearly seen that most assays are able to detect 0.1% of the impurity, the identification threshold defined by the ICH guidelines. In some cases the separation of all stereoisomers of compounds with multiple stereogenic centres has been achieved as illustrated for galantamine featuring three stereogenic carbon atoms (Figure 3). All seven distomers (undesired isomers) could be separated from the eutomer (desired isomer).20 Several methods also allow the simultaneous determination of other (achiral) related substances.


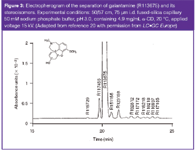
Figure 3: Electropherogram of the separation of galantamine (R113675) and its stereoisomers. Experimental conditions: 50/57 cm, 75 μm i.d. fused-silica capillary 50 mM sodium phosphate buffer, pH 3.0, containing 4.9 mg/mL α-CD, 20 °C, applied voltage 15 kV. (Adapted from reference 20 with permission from LCGC Europe)
Chiral CE methods also proved very suitable in bioanalytical studies of drugs and other analytes in physiological fluids such as urine or plasma (Table 4). In principle, direct injection of urine samples is possible, but in most cases concentration/purification steps are employed to achieve the required sensitivity. In environmental sciences CD-mediated enantioseparations were applied to the determination of pesticides and their metabolites including phenoxy acid pesticides and polychlorinated biphenyls.14


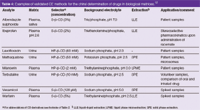
Table 4: Examples of validated CE methods for the chiral determination of drugs in biological matrices.12
What is the Performance Data Like for CD-based Methods?
CD-based enantioseparations have been developed and validated for many compounds. Typically chiral purity methods require limits of detection of around 0.1% for the undesired enantiomer to allow routine use. Chiral CE methods can routinely achieve these levels.
Upon careful validation of CE-specific parameters including buffer preparation, capillary washing procedures, etc. precision and reproducibility of the assays were comparable to HPLC methods. Compared with other chiral selectors employed in CE, CDs offer a large variety not only of derivatives but also of separation modes. Thus, extreme flexibility with regard to the experimental conditions is possible. As mentioned previously, the validation of the assay must include CDs of various commercial sources and batches during method development and the use of a system suitability test is strongly recommended. Cases have been reported where the overall performance of a CD-based chiral CE assay including the detection and quantification limits was superior to an enantioselective HPLC method.
Are There Any Reviews I Can Read?
The literature on CE enantioseparations is extensive. Numerous reviews on all aspects of chiral separations can be found in analytical journals such as Electrophoresis, Journal of Chromatography A, Journal of Separation Science, Journal of Pharmaceutical and Biomedical Analysis or Analytica Chimica Acta. Some of these journals also feature special issues dedicated to CE stereoisomer separations. Reviews dedicated to CDs in enantioseparations have been published.22–25 Summaries on chiral electromigration techniques including other chiral selectors are also available.26,27 Additionally a whole book on use of CE for chiral analysis has been published.21
Gerhard K.E. Scribas a professor at the Friedrich Schiller University, Department of Pharmaceutical and Medicinal Chemistry, Jena, Germany. His research focuses on the analysis of peptides and drugs by CE and HPLC with a special interest on stereoisomers.
Kevin Atria is an associate director in the pharmaceutical development department at GlaxoSmithKline. He is editor of CE Currents in LCGC Europe.
References
1. M.M. Rogan, D.M. Goodall and K.D. Altria, Electrophoresis, 15, 808–817 (1994).
2. Stalberg et al., Chromatographia, 41, 287–294 (1995).
3. A.C. Wren and R.C. Rowe, J. Chromatogr., 603, 235–241 (1992).
4. J.B. Vincent et al., Anal. Chem., 69, 4226–4233 (1997).
5. J.B. Vincent et al., Anal. Chem., 69, 4419–4428 (1997).
6. H. Cai, T.V. Nguyen and G. Vigh, Anal. Chem., 70, 580–589 (1998).
7. S.E. Deeb, P. Hasemann and H. Watzig, Electrophoresis, 29, 3552–3562 (2008).
8. L. Zhou et al., J. Pharm. and Biomed. Analysis, 27, 541–553 (2002).
9. M. Fillet, P. Hubert and J. Crommen, Electrophoresis, 19, 2834–2840 (1998).
10. M.J Rocheleau, Electrophoresis, 26, 2320–2329 (2005)
11. H. Atesa, D. Mangelingsa and Y. Vander Heyden, J. Pharm. and Biomed. Analysis, 48, 288–294 (2008).
12. G.K.E. Scriba, Electrophoresis, 24, 4063–4077 (2003).
13. C. Desiderio and S Fanali, J. Chromatogr. A, 807, 37–56 (1998).
14. R. Kuhn, Electrophoresis, 20, 2605–2613 (1999).
15. G. Gübitz and M.G. Schmid, in G. Subramanian (ed), Chiral Separation Techniques, 3rd Ed., Wiley, p.155–179 (2007).
16. A. Lämmerhofer, J. Chromatogr. A, 1068, 3–30 (2005).
17. K. Otsuka and S. Terabe, J. Chromatogr. A, 875, 163–178 (2000).
18. Z. El Rassi, J. Chromatogr. A, 875, 207–233 (2000).
19. K.A. Kahle and J.P. Foley, Electrophoresis, 28, 2503–2526 (2007).
20. M. Jimidar et al., LCGC Eur., 15(4), 230–242 (2002).
21. J. Hernandez-Borges et al., Electrophoresis, 26, 3799–3813 (2005).
22. S. Fanali, J. Chromatogr. A, 875, 89–122 (2000).
23. U. Schmitt, S.K. Branch and U. Holzgrabe, J. Sep. Sci., 25, 959–974 (2002).
24. Z. Juvancz, et al., Electrophoresis, 29, 1701–1712 (2008).
25. G.K.E. Scriba, J. Sep. Sci., 31, 1991–2011 (2008).
26. B. Chankvetadze, J. Chromatogr. A, 1168, 45–70 (2007).
27. G. Gübitz and M.G. Schmid, J. Chromatogr. A, 1204, 140–156 (2008).
28. B. Chankvetadze, Capillary Electrophoresis in Chiral Analysis, John Wiley & Sons, Chichester (1997).


")






; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

