The Use of Hollow Fibers in Liquid-Phase Microextraction
LCGC North America


In this installment, the subject of LPME is reviewed, with emphasis on the use of hollow fibers.
Sample preparation is a key step in chemical analysis because it is, in some cases, the slowest and most complicated step overall. Sample preparation is necessary to extract, isolate, and concentrate the analytes of interest from complex matrices into a suitable solvent because most analytical instruments cannot handle the sample matrix directly. Therefore, sample preparation has a direct impact on accuracy, precision, and quantitation limits and is often the rate-determining step of the analytical process, especially when trace determination has been the purpose.
For the isolation and concentration of analytes from aqueous matrices, the technique of liquid–liquid extraction (LLE) is still the most common extraction method even though it is time consuming and labor intensive. The technique generally is considered to be expensive since ultrapure solvents are necessary for trace analysis; it also is potentially hazardous, generates waste, and is nonenvironmentally benign and nonsustainable. Nevertheless, with improvements in analytical techniques, especially in detector sensitivity and specificity, even classical LLE has undergone a renaissance in that smaller samples can be used, which implies that extraction solvent volume can be reduced, sometimes to only a few milliliters. Now small-volume LLE can be fairly routine and can be automated.
As sample sizes continue to get smaller, techniques that provide efficient extraction are needed. Thus, a series of reduced solvent and solventless miniaturized sample preparation techniques has been developed. Many of these techniques are based on a great reduction in the volume ratio of acceptor to donor phase. In this installment of "Sample Prep Perspectives," we shall look at some of these microextraction techniques and demonstrate how the application of hollow fibers can improve the extraction efficiency, experimental robustness, and allow automation. We will focus on direct immersion hollow-fiber techniques, in which the extracting solvent is in direct or indirect contact with an aqueous sample phase (donor).
Brief Review of Microextraction Techniques
SPME and SBSE: The first successful modern microextraction technique was solid-phase microextraction (SPME) developed by the group of Pawliszyn at University of Waterloo, Ontario, Canada (1,2). This extraction technique has become the standard that other microextraction techniques have tried to emulate and outdo. The SPME technique uses a small polymer-coated fiber that is placed in solution or in the headspace of a sample for a period of time. Analytes diffuse by convection to the surface of the polymer coating until equilibrium is achieved. Once withdrawn from the sample source, in the case of gas chromatography (GC), the fiber is placed into the hot injection port and the sorbed analytes are thermally desorbed into the GC column to be separated and detected. This SPME approach was soon commercialized for both manual and automated operation. For high performance liquid chromatography (HPLC), the SPME technique never proved as successful. In this case, the analytes had to be solvent extracted (or stripped) from the polymer coating, which proved to be a slower process than thermal desorption. To overcome some of these problems, an in-tube SPME technique was developed in which analytes were sorbed by repeatedly aspirating and dispensing the sample liquid through a short piece of GC capillary whose interior walls were coated with a stationary phase (3). Then, the sorbed analytes were displaced by the HPLC solvent to the column for analysis.
A variation of the SPME technique was introduced by Baltussen and coworkers (4,5) in 1999. The stir-bar sorptive extraction technique (SBSE) involves a glass stir bar (having a magnet within) coated with polymeric sorbent (for example, polydimethylsiloxane). The available surface area of the coated stir bar is substantially higher than the surface area of an SPME coated fiber (50–250 times greater). Hence, larger amounts of analyte can be extracted and overall sensitivity is improved. A special thermal desorption unit is used to transfer sorbed analytes into the GC. The stir bar is placed in an aqueous (or other) environment, stirred for an appropriate time (usually tens of minutes), removed, dried to remove any water droplets, and placed in the thermal desorption unit for further analysis. Depending upon the octanol–water partition coefficients of the analytes, sample volume, stir-bar size, and analytical system being used (usually GC–mass spectrometry [MS]), detection limits of 1 ng/L have been obtained. The technique has been commercialized by Gerstel (Baltimore, Maryland) as the Twister extraction system.
Single-Drop Microextraction: The first attempt to emulate the SPME experiment for LLE was performed in the late 1990s (6,7). Here, a single drop (sometimes called the receiving or acceptor phase) of an immiscible extraction organic solvent (1–10 μL) was suspended from the end of a microsyringe needle in an aqueous solution (donor phase) with stirring (see Figure 1) or in a continuous flow system. This technique became known as single-drop microextraction (SDME), a subset of the general technique of liquid-phase microextraction (LPME). Common with static microextraction techniques, the process is rather slow requiring tens of minutes.


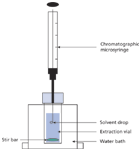
Figure 1: Schematic of the SDME apparatus.
The LPME techniques use a very small amount of organic solvent, usually only a few microliters. Due to the high volume ratio of the donor aqueous phase to the acceptor phase, analytes are concentrated during the extraction. In the case of a hanging drop (for example, SDME), once equilibrium was achieved, the drop could be retracted into the syringe barrel and injected directly into a GC, HPLC, or capillary electrophoresis (CE) instrument. The extraction of analytes from the matrix relies upon the diffusion and partition of target compounds between the solvent and aqueous phase. Therefore, the whole procedure is a process dependent upon equilibrium rather than exhaustive extraction. Parameters that accelerate the diffusion can increase the extraction efficiency, such as stirring the sample or increasing the extraction system temperature. Other parameters affecting the extraction efficiency include the volume of the solvent drop, the solvent selection, pH adjustment, salting-out effect, and so on.
Although SDME is inexpensive, simple, and efficient, the stability of the hanging drop has always been a problem. If the stirring rate was too fast, the drop could be dislodged from the syringe needle during high-speed stirring. Although selection of a syringe with a beveled needle tip, suitable solvent, and small volume of solvent (1–2 μL) can obviate this difficulty, they cannot solve the problems completely, thus limiting the development and application of the technique. Any slight solubility of the acceptor liquid in the aqueous solvent could cause the drop to change size with time, thereby affecting extraction efficiency. In addition, biological samples, such as plasma, can emulsify substantial amounts of organic solvents that also can affect the stability of the hanging drop during the extraction process.
Furthermore, drop-based LPME works best with a clean matrix because particles or bubbles in the sample matrix affect the extraction by making the drop unstable and are potentially detrimental to the analytical instrument. To protect the solvent drop, compromises must be made during the method optimization, such as shortening the equilibrium time, using a lower stirring speed, and so on, which negatively impact the extraction efficiency. In other words, the SDME method is a very good sample preconcentration technique but is generally unsuitable as a sample clean-up procedure.
Hollow-Fiber Liquid-Phase Microextraction: To circumvent the problem of drop stability, Pedersen-Bjergaard and Rasmussen (8) came up with the idea to contain the drop (or small volume of organic solvent) within the lumen of a porous hydrophobic hollow fiber, typically made of polypropylene. Thus, the drop was "protected" against mechanical disturbance within the confines of the fiber. The technique has been referred to as hollow-fiber liquid-phase microextraction (HF-LPME). With the presence of the hollow fiber, extractions could be performed in either a two-phase mode or a three-phase mode. The two-phase mode consists of the organic solvent (a few microliters) in the lumen as well as being immobilized in the wall pores of the hollow fiber. The extracting solvent (acceptor) is not in direct contact with the sample solution because the porous membrane of the hollow fiber serves as a barrier to the aqueous sample. The porous hollow fiber also could serve as a three-phase extraction system in which the hollow fiber is first treated with organic solvent that provides a supported liquid membrane (SLM) with the immiscible solvent filling the pores of the membrane. An acceptor solution, usually aqueous, is placed inside the lumen of the hollow fiber. Thus, the three-phase system could be used to perform liquid–liquid–liquid microextraction (LLLME). The SLM technique was pioneered by Audunsson (9) and popularized by Jonsson and Mathiasson (10). Later, we will get into more details of the workings of HF-LPME.
Dispersive liquid–liquid microextraction: A technique called dispersive liquid–liquid microextraction (DLLME) is based upon a ternary component solvent system (11,12). In this method, also useful for extractions of analytes from aqueous media, a small volume of extracting solvent (higher density than water and about 1–3% of the total volume of extracting mixture) and a disperser solvent (miscible with extracting solvent and aqueous sample solvent) are mixed together and are rapidly injected into the aqueous sample. A cloudy mixture is formed due to the tiny droplets of immiscible extracting solvent in the aqueous environment. Due to the high surface area of these extracting solvent droplets, analyte extraction is quite efficient and extremely rapid. The extracting solvent containing analyte is then centrifuged and the sedimented phase collected for direct analysis or further workup.
Solidified floating drop microextraction: Solidified floating drop microextraction (SFODME) is a technique similar to DLLME in that a small volume of immiscible organic solvent with a melting point near room temperature is floated on the surface of an aqueous sample containing analyte (13). The phases are stirred for a period of time and the mixture transferred to an ice bath. The organic solvent is solidified, removed, and placed in a vial where it melts, thereby releasing the analyte for further analysis. This approach of DLLME allows the use of an extracting solvent that is less dense than water.
HF-LPME
Hollow fibers are used in a wide variety of applications, the major one being their use for wastewater treatment but in the laboratory, they have been used for microfiltration and reverse osmosis. They are made of organic polymers such as polypropylene, polyethersulfone, polyester, and other materials and also can be constructed from inorganic materials such as titania and zirconia. The hollow fibers are relatively inexpensive and can be considered for single use only; in addition, the materials are recyclable. To remove potential contaminants, prior to use in any analytical application, hollow fibers should be rinsed in acetone or other compatible solvent several times, then dried at room temperature.
The apparatus for static HF-LPME is illustrated in Figure 2. Rather than the spherical configuration of a drop, the HF-LPME configuration provides a rod-like shape of extracting solvent. The rod-like configuration increases the solvent surface area because for the same volume of liquid, the surface area of a sphere is lower. The contact area between sample solution and extracting solution is thus much more substantial than if the solvent were spherically shaped. The direct benefit of the change in solvent configuration is better extraction efficiency. Another significant benefit is that higher stirring speeds can be applied during the extraction procedure, because the solvent is protected by the hollow fiber and its stability is enhanced. In addition, the disposable nature of the hollow fiber eliminates the possibility of sample carryover and ensures high reproducibility; and the pores in the walls of the hollow fiber cause it to display some selectivity by preventing the extraction of macromolecules, such as proteins, and particles from the sample matrix. Therefore, HF-LPME is no longer just a preconcentration technique. It also can provide simultaneous sample clean up, and it is suitable for use with complex sample matrices.

Figure 2: Schematic of the HF-LPME apparatus.
The selection of organic solvent used to fill in the pores of hollow fiber is a critical parameter in the success of the method. There are several requirements for the organic solvent. Firstly, it should be immobilized easily in the hollow-fiber pores, and it should be nonvolatile. Typically, the hollow fibers used in most HF-LPME experiments are made of hydrophobic polypropylene, and the solvent selected should have good affinity for it to prevent solvent loss during the extraction, and also to achieve stable solvent impregnation. Secondly, the solvent should be immiscible with water as it serves as a barrier to the aqueous sample. Lastly, the solubility of the analyte of interest in the solvent should be higher than that in the aqueous sample matrix. Usually a relatively nonpolar, high viscosity solvent will meet those requirements, and typical solvents used include 1-octanol, n-hexyl ether, undecane, toluene, and so forth. However, these relatively nonpolar solvents usually have low solubility for the polar compounds, thus negatively impacting the extraction efficiency for polar compounds. Recently, ionic liquids have been used as solvents for HF-LPME applications (14–17). Ionic liquids are polar and nonvolatile, but have been demonstrated to generate stable solvent impregnation. Figure 3 shows scanning election microscopic images of the inner surface of the hollow fiber before and after ionic liquid impregnation (7). By comparing the two photographs, it is obvious that the ionic liquid was immobilized in the hollow-fiber pores effectively and an ionic liquid membrane for extraction formed. The advantages of using an ionic liquid as the extraction solvent include high affinity for polar compounds and compound-dependent selectivity. These specific features of ionic liquids have broadened the applications of HF-LPME to polar compound analysis.

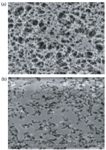
Figure 3: Scanning election microscopic images of the inner surface of the hollow fiber (a) before and (b) after ionic liquid impregnation.
The HF-LPME procedure is simple and includes just a few steps (see Figure 2). Before extraction, the hollow fiber (typically, 1.5–10 cm in length, 200-μm thick, 600-μm i.d. with a 0.2-μm wall pore size), usually sealed at one end and attached to the syringe needle on the other end, is soaked into the immiscible solvent that results in the immobilization of the solvent into its wall pores. The solvent forms a thin layer within the wall of the hollow fiber. A microsyringe usually is used to support the hollow fiber and also to introduce the acceptor phase into its lumen. The hollow fiber with added acceptor solution is then placed into a sample vial filled with the aqueous sample, usually with a volume of several milliliters. To speed up the extraction, the sample is stirred extensively. The analytes are extracted from the aqueous sample through the solvent layer in the pores of the hollow fiber, then into the acceptor phase inside its lumen. After a certain time of extraction to achieve equilibrium (often in the range of 10–30 min), the acceptor phase is withdrawn into the microsyringe and then injected directly into the instrument for analysis.
Depending upon the mode, the lumen of the hollow fiber can be filled with several microliters of the same solvent (two-phase mode) or an aqueous acceptor phase (three-phase mode). In two-phase HF-LPME, organic solvent impregnated in the hollow-fiber segment is exposed as a solvent rod in a stirred aqueous sample. Nonpolar analytes in the aqueous sample then can be extracted largely through the hollow fiber into the acceptor solvent by diffusion. Because the solvent used is usually GC-amenable, the extracted sample can be then injected directly into the GC system for analysis. This two-phase HF-LPME is more suitable for the extraction of relatively hydrophobic analytes (17–19).
The three-phase mode LLLME is sometimes referred to as an LPME with back extraction (LPME-BE). In this mode, both donor and acceptor phases are aqueous-based, and the third solvent is an organic phase immobilized into the pores of the hollow fiber, which serves as a barrier to separate the two aqueous phases. During the extraction, the target analytes are extracted from the donor aqueous phase through the thin layer of organic solvent within a hollow-fiber wall and then into the acceptor aqueous phase. Because the acceptor phase usually is aqueous, the extracts can be analyzed by HPLC or CE. This method is applied for ionic or ionizable compounds, and mass transfer is driven largely by the differences between the donor and acceptor aqueous phases, such as pH adjustment, salting out effect in donor phase, or addition of proper acceptor reagent like ion-pair complex reagent for analytes (20–22).
Using the SLM principle, Figure 4 represents a typical example of how the LLLME works for an organic acid analyte in aqueous sample solution. If the pH of the donor solution is below the pKa of the acid, it will be nonionized and will diffuse and partition into the organic phase within the pores of the hollow fiber. If the acceptor aqueous solution in the lumen of the hollow fiber is at a pH much higher than the acid's pKa, the neutral acid will now be ionized as the carboxylate ion and will remain in the acceptor solution, preferring not to diffuse back into the organic layer of the hollow fiber.


Figure 4: LLLME of organic acid from an aqueous sample.
As the phase ratio between the donor phase (aqueous sample) and the acceptor phase (solvent or second aqueous phase) is very large (>100:1), the analytes are concentrated significantly, up to several hundreds times enrichment factor, in the acceptor phase. As a result, the detection limit of the analytical method is improved greatly. Therefore, lower detection limits (lower parts-per-billion level or even sub-part-per-billion level) can be achieved easily, even with relatively low sensitivity detectors such as a UV detector.
Variations of HF-LPME
Since the development of HF-LPME, there have been many modifications and variations to improve on the extraction efficiency, selectivity, and interfacing to the analytical instruments. Table I provides a brief description of some of these variations and provides references for those who wish to learn more about them. One variation (not covered in Table I) is the dynamic HF-LPME mode (23,24). Here, the syringe with the hollow fiber attached to its needle is clamped onto a syringe pump, and the syringe plunger can be pulled back and forth by the syringe pump. During the extraction, a small volume of aqueous sample is automatically pulled in and out of the hollow fiber repeatedly by the syringe plunger. When the aqueous sample is withdrawn, a thin film of organic solvent is established on the hollow-fiber wall and the analytes are extracted from the sample segment. During sample dischargement, this thin film recombines with the bulk solvent in the syringe, and the extracted analytes are trapped in the bulk organic phase. After a prescribed number of repeated cycles, a portion of the bulk extract can be retrieved for instrumental analysis. Dynamic HF-LPME improves extraction speed (on the order of a few minutes) compared with static systems, but the operation in dynamic mode is more complicated. By automation, the dynamic process can be quite reproducible as well as provide high enrichment factors.

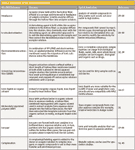
Table I: Variations of the HF-LPME technique
Selected Applications of HF-LPME
Two-Phase Extraction: For analytes that have a high water–octanol partition coefficient (Kow), a simple two-phase HF-LPME setup can be used. In this approach, the same organic acceptor phase is contained in both the hollow-fiber pores as well as in the lumen. Because the acceptor phase is usually organic, this technique is interfaced to GC more easily. Li and coworkers (41) applied HF-LPME as a sample preparation technique to investigate oil spills. After weather simulating a typical crude oil spill under laboratory conditions at various exposure times ranging from 0.1 to 72 h, for sample preparation, the scientists used the same configuration of HF-LPME device as the originators (8), but substituted a polyvinylidene difluoride hollow fiber (500-μm i.d., 150-μm wall thickness, 0.2-μm pore size, 2-cm length) instead. The membrane was sonicated in dichloromethane three times to remove any contaminants and dried. The hollow fiber was immersed in n-hexane for 5 min. Next, 3 μL of water was used to displace any hexane from the lumen followed by 4 μL of n-hexane to serve as the acceptor phase. The total aqueous sample volume was 4 mL. Extraction took place for 30 min with a 700-rpm magnetic stirrer used to agitate the sample. At the end of the 30-min extraction, the 4 μL of hexane was withdrawn from the hollow fiber and, after dilution with n-hexane, placed in a sample vial for analysis by GC, GC–MS and GC–isotope ratio (IR) MS. The authors compared the HF-LPME results to those obtained with conventional LLE on the weathered crude oil samples and confirmed that they achieved the same results. However, there were some marked advantages of the HF-LPME technique. Because the hollow fiber is unaffected by large molecules and suspended solid particles, an extra clean-up step was not required and the total experimental time was less. Although not pertinent to this study, in which sample amount was not a limitation, the advantage of higher enrichment noted with HF-LPME over LLE was not taken into account.
Three-Phase Extraction: The most popular form of LLLME involves an aqueous acceptor phase that is more amenable to interfacing with HPLC and CE techniques. Zhao and colleagues (42) demonstrated that aromatic amines in water samples can be extracted (with stirring) and concentrated using a three-phase hollow-fiber extraction system. The target amines were extracted from a 4-mL aqueous sample (donor) that was adjusted to pH 13 with 0.1 M sodium hydroxide. At this pH, the amines were in an unionized state and thus could partition into the organic solvent immobilized in the pores of the hollow fiber (1.5-cm length and 600-μm i.d.). The solvent used was di-n-hexyl ether. The acceptor solution was a 4-μL acidic solution. In this solution, the amine was now protonated and therefore could not be extracted back into the organic layer. Some of the variables in the extraction procedure were investigated. For example, a study of the extraction time revealed that at a 1000-rpm stir rate, 20 min is required to establish equilibrium. A slower stir rate required a longer time to achieve equilibrium. Phase ratios of donor and acceptor solutions were varied from 2000:1 to 800:1. For a 4-mL donor phase volume, the best extraction efficiency occurred when 4 μL of 0.1 M hydrochloric acid was used as the acceptor solution (for example, with a 1000:1 phase ratio). A study of the use of salting out in the donor phase to aid extraction efficiency showed that the addition of 20% sodium chloride was best. A small amount of acetone (2% by volume) was added to the donor solution as it was found to facilitate analyte transfer into the organic solvent in the pores of the hollow fiber. Furthermore, extraction efficiency was improved by the addition of 18-crown-6 ether into the acceptor phase since it complexed with the protonated aromatic amines and increased the efficiency factors by as much as 30%. For quantitation, interday % RSDs for the four amines studied were in the range of 3.8–4.8% with limits of detection of 0.05–0.10 μg/L with good linearity. Similar to other LPME techniques, enrichment factors were quite large, ranging from 240 up to 470. Because humic acids are always present in naturally occurring waters, a study on the addition of humic acid up to 200 mg/L showed that there was little effect on the extraction efficiency. A study of spiked real-water samples demonstrated the LLLME technique could provide excellent trace enrichment of aromatic amines.
HF-LPME for inorganic species: Most applications of HF-LPME have been demonstrated for organic species. However, by the use of complexation, inorganic species can be enriched from aqueous solution. In the work of Li and Hu (43), vanadium (IV/V) species could be separated and determined using ammonium pyrrolidinecarbodithioate as a chelating reagent and using carbon tetrachloride as the extraction solvent. Measurement of the extracted chelated metals was made by inductively coupled plasma–optical emission spectroscopy (ICP-OES). Speciation studies were performed by selectively masking one of the vanadium complexes. In environmental water samples, enrichment factors of 74 and detection limits of 86 pg/mL for vanadium(IV) and 71 pg/mL for vanadium(V) were noted. Recently, Dadfarnia and Haji Shabani (44) reviewed the determination of trace level concentrations of metals by LPME including HF-LPME.
Conclusion
Miniaturized sample preparation procedures are gaining attention and momentum. LPME is characterized by simplicity — any laboratory can use it with a basic apparatus (for example, SDME). HF-LPME is particularly rugged and can be used for significantly dirty environmental matrices. The LLLME mode is quite versatile and can be applied to HPLC and CE because extracts are aqueous-based. Using a variety of organic solvents, including ionic liquids, and solvent-mixtures allows the LPME techniques to provide a high degree of selectivity. The extraction approach is green and chemically sustainable and has great potential for portability and field applicability. The technique can be automated for improved throughput and precision. More recently, with the advent of electromembrane extraction in which electrical potential is used as a driving force, another parameter is available to further enhance extraction.
Ronald E. Majors Ronald E. Majors "Sample Prep Perspectives" Editor Ronald E. Majors is Senior Scientist, Columns and Supplies Division, Agilent Technologies, Wilmington, Delaware, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.


Limian Zhao Limian Zhao received her B.Sc. in Chemistry in 1997 from Lanzhou University, P.R. China. She then obtained her M.Sc. in Analytical Chemistry from National University of Singapore, and M.Sc. in Food Chemistry from Rutgers, the State University of New Jersey, respectively. She is currently employed as an application chemist at Agilent Technologies in Wilmington, Delaware.


Hian Kee Lee Hian Kee Lee is Professor with the Department of Chemistry, National University of Singapore. He obtained the B.Sc. (Honors) and Ph.D. degrees from the University of Canterbury, New Zealand. His main research interest is in the development of microscale solvent-minimized sample preparation techniques.
References
(1) C.L. Arthur and J. Pawliszyn, Anal. Chem. 66, 2145–2148 (1990).
(2) C.L. Arthur, D.W. Potter, K.D. Buchholz, S. Motlagh, and J. Pawliszyn, LCGC North America 10(9), 656–661 (1992).
(3) R. Eisert and J. Pawliszyn, Anal. Chem. 69(16) 3140–3147 (1997).
(4) E. Baltussen, P. Sandra, F. David, and C.A. Cramers, J. Microcol. Sep. 11, 737 (1999).
(5) F. David, B. Tienport, and P. Sandra, LCGC North America 21(2), 108–118 (2003).
(6) H. Liu and P.K. Dasgupta, Anal. Chem. 68, 1817–1821 (1996).
(7) M.A. Jeanott and F.F. Cantwell, Anal. Chem. 68, 2236–2240 (1996).
(8) S. Pedersen-Bjergaard and K.E. Rasmussen, Anal. Chem. 71, 2650–2656 (1999).
(9) G.A. Audunsson, Anal. Chem. 58, 2714–2723 (1986).
(10) J.A. Jonsson and L. Mathiasson, J. Chromatogr., A 902, 205–225 (2000).
(11) D. M. Razaee, Y. Assadi, M.R. Milani Hosseini, E. Aghaee, F. Ahmadi, and S. Berijani, J. Chromatogr., A 1116, 1–9 (2006).
(12) R.E. Majors, LCGC North America 26(12), 1158–1166 (2008).
(13) E.M.R. Khalili Zanjani, Y. Yamini, S. Shariati, and J.A. Jonsson, Anal. Chim. Acta 585, 286–293 (2007).
(14) U. Tao, J.-F. Liu, X.-L. Hu, H.-C. Li, T. Wang, and G.-B. Jiang, J. Chromatogr., A 1216, 6259–6266 (2009).
(15) J.-F. Peng, J.-F. Liu, X.-L. Hu, and G.-B. Jiang, J. Chromatogr., A 1139, 165–170 (2007).
(16) J.-F. Liu, G.-B. Jiang, Y.-Q. Cai, Q.-X Zhou, and J.-T. Hu, Anal. Chem. 75, 5870–5876 (2003).
(17) J.-F.Liu, Y.-G. Chi, G.-B. Jiang, C. Tai, J.-F. Peng, and J.-T. Hu., J. Chromatogr., A 1026(1–2), 143–147 (2004).
(18) T.S. Ho, S. Pedersen-Bjergaard, and K.E. Rasmussen, J. Chromatogr., A 963 (1–2), 3–17 (2002).
(19) X. Jiang, S.Y. Oh, and H.K. Lee, Anal. Chem. 77, 1689–1695 (2005).
(20) A. Saleh, Y. Yamini, M. Faraji, S. Shariati, and M. Rezaee, J. Chromatogr., B 877, 1758–1764 (2009).
(21) L. Zhao and H.K. Lee, J. Chromatogr., A 931(1–2), 95–105 (2001).
(22) L. Xia, B. Hu, and Y. Wu, J. Chromatogr., A 1173(1–2), 44–51 (2007).
(23) Y. Wu and B. Hu, J. Chromatogr., A 1216(45), 7657–7663 (2009).
(24) Y. He and H.K. Lee, Anal. Chem. 69, 4634–4640 (1997).
(25) L. Zhao and H.K. Lee, Anal. Chem. 74, 2486–2492 (2002).
(26) X. Jiang, C. Basheer, J. Zhang, and H.K. Lee, J. Chromatogr., A 1087, 289 (2005).
(27) L. Meng, X. Liu, B. Wang, G. Shen, Z. Wang, and M. Guo, J. Chromatogr., B 877, 3645–3651 (2009).
(28) M.I. Carpinteiro, I. Rodriguez, R. Cela, and M. Ramil, J. Chromatogr., A 1216, 2825–2831 (2009).
(29) H.S.N. Lee, M.T. Sng, C. Basheer, and H.K. Lee, J. Chromatogr., A 1148, 8–15 (2007).
(30) K.-J. Chia and S.-D. Huang, J. Chromatogr., A 1119, 161 (2006).
(31) K.E. Kramer and A.R.J. Andrews, J. Chromatogr., B 760, 27 (2001).
(32) C. Basheer, A. Jayaraman, M.K. Kee, S. Valiyaveettil, and H.K. Lee, J. Chromatogr., A 1100, 137–143 (2005).
(33) S. Pedersen-Bjergaard and K.E. Rasmussen, J. Chromatogr., A 1109, 183–190 (2006).
(34) A. Gjelstad, LCGC North America 28(2), 92–112 (2010).
(35) X. Jiang and H.K. Lee, Anal. Chem. 76, 5591–5596 (2004).
(36) Y. Hu, Y. Wang, Y. Hu, and G. Li, J. Chromatogr., A 1216, 8304–8311 (2009).
(37) J. Wu and H.K. Lee, J. Chromatogr., A 1133, 13–20 (2006).
(38) J. Wu and H.K. Lee, Anal. Chem. 78, 7292–7301 (2006).
(39) H.-C. Chen, W.-T. Chen, and W.-H. Ding, Talanta 79(2), 442–445 (2009).
(40) H.Jiang, B.Hu, B.Chen, and W. Zu., Spectrochim. Acta B 63, 770–776 (2008).
(41) Y. Li, Y. Xiong, J. Fang, L. Wang, and Q. Liang, J. Chromatogr., A 1216, 6155–6161 (2009).
(42) L. Zhao, L. Zhu, and H.K. Lee, J. Chromatogr., A 963, 239–248 (2002).
(43) L. Li and B. Hu, Talanta 72, 472–479 (2007).
(44) S. Dadfarnia and A.M. Haji Shabani, Anal. Chim. Acta 658, 107–119 (2010).









Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.

