Understanding the Lifecycle Approach for Analytical Procedures
LCGC North America
A key component for data integrity is having accurate and precise analytical procedures that are validated for intended use. Changes in the way that procedures are specified, developed, validated, and operated are coming. Here is what you need to know.
Accurate analytical analysis requires robust and validated analytical procedures. Change is underway in the approach to developing, validating, and using analytical procedures. Are you ready for the changes coming your way?
All analytical procedures should be fit for their intended use with appropriate measurement uncertainty (precision and accuracy), selectivity, and sensitivity. In this installment of “Data Integrity Focus,” we look at the impact of an analytical procedure on the integrity of data produced in regulated good manufacturing practices (GMP) and good laboratory practices (GLP) laboratories. Within the framework of the Data Integrity Model (1, 2), there is the right analytical procedure for the job at Level 2. The use of an accurate procedure is built on the foundation layer of data governance with management leadership, quality culture, procedures for data integrity, and training. This is applied to getting the right analytical instrument and application software that are qualified and validated respectively. Both levels now need to be applied to the development, validation, and use of any analytical procedure.
Analytical Procedure or Method?
You will notice that the title of this article uses the term analytical procedure, and not analytical method. The reason is that an analytical procedure covers all stages from sampling, transport, storage, preparation, analysis, interpretation of data, calculation of the reportable result, and reporting. An analytical method is a subset of this, and is typically interpreted as the instrumental analysis phase. After a discussion of the applicable regulations and guidance, I will focus on the analytical method portion of a procedure. After all, this is LCGC!
GMP Regulatory Requirements for Analytical Procedures
In 21 CFR 211.194(a), there is the following requirement for analytical methods used in pharmaceutical analysis:
(a) Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays,
as follows: …..
(b) A statement of each method used in the testing of the sample.
The statement shall indicate the location of data that establish that the methods used in the testing of the sample meet proper standards of accuracy and reliability as applied to the product tested…..
The suitability of all testing methods used shall be verified under actual conditions of use (3).
What does this mean in practice? Any laboratory must know where the validation was carried out so that an inspector can access the data plus any method transfer protocol that was performed with the associated report to show that the procedure works in a specific laboratory. This interpretation is mirrored in EU GMP Chapter 6 on Quality Control, where clause 6.15 states:
Testing methods should be validated.
A laboratory that is using a testing method and which did not perform the original validation, should verify the appropriateness of the testing method.
All testing operations described in the marketing authorization or technical dossier should be carried out according to the approved methods (4).
Reinforcing the European requirement, there is also EU GMP Annex 15 on Qualification and Validation, a very generic set of requirements covering all possible processes and equipment, where Section 9.1 notes for test methods:
All analytical test methods used in qualification, validation or cleaning exercises should be validated with an appropriate detection and quantification limit, where necessary, as defined in Chapter 6 of the EudraLex, Volume 4, Part I (5).
However, these regulations give broad direction, but not much detail. What do we need to do to validate an analytical procedure or test method?
GMP Regulatory Guidance for Validation
Currently in GMP, there is ICH Q2(R1) for method validation (6) that outlines the requirements for method validation for quality control (QC) testing. The emphasis in the document is mainly on chromatographic methods of analysis with parameters such as repeatability, intermediate precision, limits of quantification (LOQ), and limits of detection (LOD). There is no mention of method development in the guidance. However, there is an almost ritualistic approach to interpreting ICH Q2(R1): “If it says it, do it.”
Therefore, we can find the stupid situation when validating a method for an assay of active component between say 90 and 110% of label claim, that the method also includes determination of LOQ and LOD. Why determine such parameters when the method will never be used near them? It is in ICH Q2(R1)” is always the answer. This is mirrored in EU GMP Annex 15, where at first reading all analytical procedures appear to require LOQ and LOD determination. However, the requirement does say “as appropriate.” Does anyone ever engage the brain and think in these situations?
In 2000, the FDA issued a draft guidance for industry on Analytical Procedures and Methods Validation (7) that outlined the FDA expectations for validation. The main problem is that this guidance did not address one of the most critical stages of the whole process: method development. In 2015, the FDA replaced the 2000 draft guidance with yet another draft guidance entitled “Analytical Procedures and Method Validation for Drugs and Biologics” (8), where there is a little, but insufficient, section on method development.
Bioanalytical Method Validation Guidances
In the bioanalysis field, there are guidances issued by the EMA and FDA. The European Medicines Agency Guideline on Bioanalytical Methods Validation from 2011 states in Section 4.1 (9):
A full method validation should be performed for any analytical method whether new or based
upon literature.
The main objective of method validation is to demonstrate the reliability of a particular method for the determination of an analyte concentration in a specific biological matrix, such as blood, serum, plasma, urine, or saliva. Moreover, if an anticoagulant is used, validation should be performed using the same anticoagulant as for the study samples. Generally, a full validation should be performed for each species and matrix concerned.
The final version of the FDA Bioanalytical Methods Validation guidance for industry in 2018 contains in the section on Guiding Principles the following selected statements (10):
The purpose of bioanalytical method development is to define the design, operating conditions, limitations, and suitability of the method for its intended purpose and to ensure that the method is optimized for validation.
Before the development of a bioanalytical method, the sponsor should understand the analyte of interest (determine the physicochemical properties of the drug, in vitro and in vivo metabolism, and protein binding) and consider aspects of any prior analytical methods that may be applicable.
Method development involves optimizing the procedures and conditions involved with extracting and detecting the analyte. Bioanalytical method development does not require extensive record keeping
or notation…..
While this FDA guidance has started to include method development, it notes that documentation of this work does not need to be extensive. As we shall see later, this is the wrong approach to take, as method development is the single most important phase of an analytical procedure life cycle. Get this right, and the validation and operation of the method are easier to handle than a rushed development and validation. If a rushed approach is taken, then the analysts using the method pick up the tab with variable results and out-of-specification investigations.
In 2019, ICH M10 on Bioanalytical Method Validation reached step 2b and was issued for public consultation. Out of 60 pages, method development receives a scant half a page mention along the lines of FDA and EMA above and noting that:
Bioanalytical method development does not require extensive record keeping or notation (11).
All the emphasis is on the validation, rather then the development of the assay. As we shall see, this is not the smartest approach, especially the majority of bioanalytical methods can be measuring analytes in biological matrices at the LOQ of the method. You really need to know what factors you need to control in the method, rather than hoping for the best.
Traditional View of Development, Validation and Use
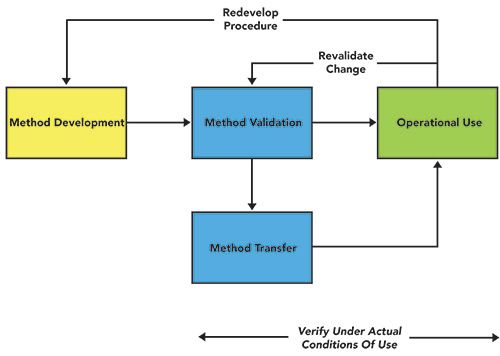
Continuing this theme, the traditional view of analytical method development, validation, and use is shown in Figure 1. The main emphasis is on a rapid method development phase and validation by an analytical development group. This is followed by a formal transfer to a quality control group to demonstrate that the method (possibly) works in their laboratory and then operational use by the QC staff. If changes are required, these need to be validated, and a method may need to be redeveloped in light of experience with use. As we shall discuss at the end of this article, most methods are regulated for the pharmaceutical laboratory, and need regulatory approval for any major change. How can this be simplified?


Figure 1: A traditional view of analytical method development, validation, and use.
USP is Changing to a Lifecycle Approach
For 10 years, USP expert panels and committees have been publishing stimuli articles on analytical procedure lifecycle management (APLM). This new approach comes from the FDA’s updated guidance on process validation that took a lifecycle approach to the topic, rather than “three validation batches and all is good.” In addition to the “Stimuli to the Revision Process” articles published in Pharmacopoeial Forum, there is also a draft USP <1220> on Analytical Procedure Lifecycle Management issued for public comment in 2017 (12). At the end of this year, a revised draft of USP <1220> is expected to be published for comment.
The principles outlined in the current draft USP <1220> are a Quality by Design (QbD) approach to method development and validation (12) that is intended to deliver more robust analytical procedures. There is greater emphasis on the earlier phases of the lifecycle of an analytical procedure, such as defining the procedure specification in an Analytical Target Profile or ATP.
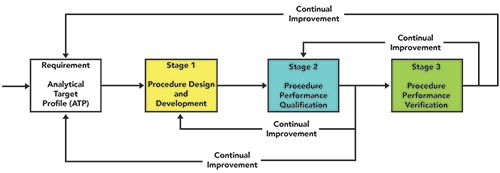
The overall process is shown in Figure 2. Although the USP is focused primarily on compendial analytical procedures, the sound scientific principles outlined in the draft USP <1220> are, in my view, applicable to bioanalytical methods as well. Shown also in the figure are the feedback loops from stage 3 to stage 2 and from stage 2 to stage 1, as well as to the ATP, representing continual improvement of the procedure. The key is continual improvement, as the pharmaceutical industry is regulated and some procedures that are part of a registration dossier might need to be modified under change control.

Figure 2: Proposed USP <1220> process for analytical procedure lifecycle management.
Stages of the Analytical Procedure Lifecycle
The lifecycle of analytical procedure advocated by USP <1220> in Figure 2 consists of three stages:
- Procedure Design and Development (method development) derived from the ATP
- Procedure Performance Qualification (method validation)
- Procedure Performance Verification (Ongoing assessment of the procedure performance).
We are not very good at method development or monitoring performance of an analytical procedure in use. This new approach aims to provide a sound scientific basis throughout the whole analytical procedure lifecycle. We will discuss each stage of the lifecycle in overview; for a more detailed understanding of the USP <1220> process and best practices in analytical procedure validation, the reader is referred to the book by Ermer and Nethercote (13).
Define the Analytical Target Profile
First, we need to define what are the objectives of the procedure and this is achieved by writing an Analytical Target Profile (ATP), as shown in Figure 2. An ATP should be considered the specification or intended use for any procedure. This term was developed by a US and EU pharmaceutical industry working group on Analytical Design Space and Quality by Design of Analytical Procedures, and has been incorporated by the USP into two Stimuli to the Revision Process articles on the ATP, as well as the draft USP general chapter <1220> (12–15).
The Analytical Target Profile (ATP) for an analytical procedure is a predefined objective of a method that encapsulates the overall quality attributes required of the method, including:
- sample to be tested
- matrix that the analyte will be measured in
- analyte(s) to be measured
- range over which the analyte(s) are to be measured for the reportable result
- quality attributes such as selectivity and precision and accuracy of the whole procedure or total measurement uncertainty (TMU).
This is the core of the lifecycle approach, as it defines the high-level objectives with no mention of any analytical technique used to meet the ATP as this could bias the analytical approach.
An example ATP could be:
To quantify analyte X over a range between a% and b% (or whatever units are appropriate) with X% RSD precision and Y% bias in a matrix of Z (or in the presence of Z).
This means that the requirements for an analytical procedure are defined before any practical work begins, or even an appropriate analytical technique has been selected. It provides the method developer with an explicit statement of what the procedure should achieve. This is a documented definition, and can be referred to during development of the procedure or revised as knowledge is gained.
Stage 1: Procedure Design and Development
This is most important part of an analytical procedure lifecycle, but it is missing from or minimal in the current regulatory guidance documents described above. Knowing how sampling, transport, storage, instrumental analysis parameters, and interpretation of data impact the reportable value is vitally important to reducing analysis variability, and hence out-of-specification (OOS) results. The aim of a Quality by Design (QbD) approach is a well understood, controlled, and characterized analytical procedure, and this begins with the design and development of the procedure.
Knowledge Gathering
From the ATP we need to gather information and knowledge to begin the initial procedure design, such as:
- chemical information about the analytes of interest, such as structure, solubility, and stability (if known)
- literature search (if a known analyte) or discussions with medicinal chemists (if a new molecular entity, or NME).
From this knowledge, coupled with the ATP, the most appropriate procedure including the measurement technology can be derived, such as:
- type of procedure (for example, assay or impurity) in an active pharmaceutical product or determination of an NME in animal or
- human plasma.
- sampling strategy, such as the sample amount or volume required, how the sample will be taken, any precautions required to stabilize the analyte in the sample, and other factors
- design of the sample preparation process to present the sample to
- the instrument
- whether there is any need to derivatize
- the analyte to enhance detection characteristics
- appropriate analytical technique to use based upon the ATP and the chemical structure of the analyte (including, but not limited to, LC-MS, LC-UV, and GC-FID)
- an outline of separation needs based on previous analytical methods with analytes of similar chemical structure,
- if appropriate.
In addition, business factors such as time for the analysis and cost should be considered when developing a method. Quicker is better, provided that the ATP is met, and UHPLC may be a better alternative to conventional HPLC, as an example.
Initial Design of the Analytical Procedure
Assuming that we are dealing with a liquid chromatographic analysis, solid samples need to be prepared so that a liquid extract can be introduced into the chromatograph for analysis. Development of the sampling, sample preparation, and separation should proceed in tandem and iteratively as shown in Figure 3. Some considerations for this phase of the development, covering all sample types and concentration or amount ranges defined in the ATP are:
- How much sample is required to achieve the ATP?
- Does the sample have to be dissolved, homogenized, sonicated, or crushed before sample preparation can begin?
- Does the analyte require derivatization either to stabilize the compound, or to enhance limits of detection
- or quantification?
- Screening experiments are run to see how the analytes run on a variety of columns and mobile phases of varying composition of organic modified and pH value of the aqueous buffers. It is important to note here that the KISS (Keep It Simple, Stupid) principle applies here. Don’t overcomplicate a method, as it will usually need to be established in one or more other laboratories, and unnecessary complexity makes method transfer more difficult.

Figure 3: Method development workflow for an HPLC procedure (13).
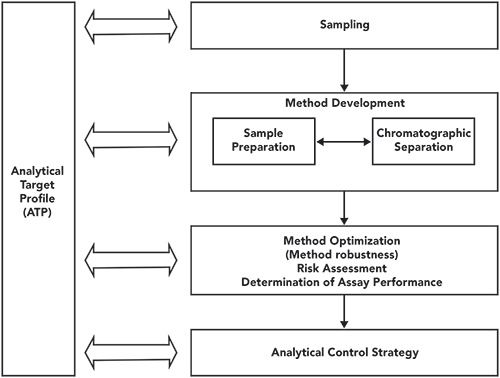
A better approach for method screening is to automate it, using method development software to design and execute experiments using a statistical design (for example, factorial design such as Plackett-Burman). This is a more expensive option, but it will produce design space maps for optimum separation much faster than a manual approach. These design space maps provide the basis for a robust separation as the factors controlling the separation can be more easily identified and the optimum separation to meet the ATP can be predicted and then confirmed by experiment.
The overarching principle in method development and optimization is to keep the method as simple as possible to achieve the ATP requirements. For example, a commonly available column and simple mobile phase preparation should be the starting point for most separations, depending, of course, on the type of analyte involved. Use isocratic elution to achieve the ATP rather than a gradient, as the latter will increase the overall analysis time.
Risk Assessment and Management
Management of risk is a key element in the analytical procedure lifecycle approach. This involves identifying and then controlling factors that can have a significant impact on the performance of the separation. Such factors may be:
- pH value of the aqueous buffer or proportion of organic modifier used in an LC mobile phase
- type or dimensions of the column used
- autosampler and column temperature
- impact of light during sampling or sample preparation.
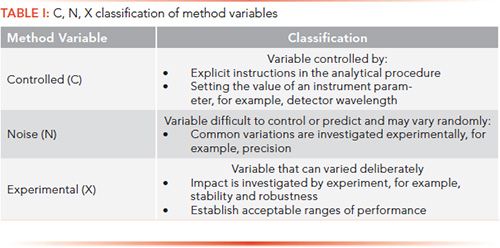
A formal risk assessment can be undertaken, such as Failure Mode Effects Analysis (FMEA), to identify the risk with the highest impact (13). The aim of risk assessment is to either mitigate or eliminate the risk posed by variables in the sample preparation, instrumental analysis, or operating practices. Method variables can be classified as controlled, noise, or experimental (C, N, or X), as shown in Table I. A discussion of how these variables are investigated is outside the scope of this article, and the reader is referred to Ermer and Nethercote’s book for more details (13).

When key variables have been identified, then robustness studies can be started to understand the impact of each one on the overall analytical procedure. There will be a study design for robustness experiments, and the results will be examined statistically. The aim is to identify the acceptable range of each key variable; the greater the range means that the method is more flexible. Again, please see Ermer and Nethercote for more information about this approach (13).
Analytical Control Strategy: Identifying and Controlling Risk Parameters
The analytical control strategy for each procedure is based on the outcome of the risk assessment and, where appropriate, in combination with the robustness studies. This should provide a list of method parameters and variables that have significant impact on the method, and its performance as well as what to avoid when executing the analytical procedure. The outcome is the establishment of controls for critical parameters, such as how to perform a specific task with sufficient detail to ensure consistent performance, the type of integration, conditions in the procedure that have significant effects, or steps to avoid certain situations where outside variables (light, for example) can affect the stability of the analyte.
The outcome of the analytical control strategy is to have a set of instructions that are explicit and unambiguous when executing the procedure, such as:
- how to sample and the required sample size
- specification of sample containers, transport conditions to the laboratory, and storage conditions
- preparation of the sample for analysis
- preparation of reference standard solutions and mobile phases
- performance of the analysis, as well as integration and interpretation of data
- calibration method used
- identification of the system suitability test (SST) parameters to be used, and determination of the acceptance criteria for each one.
Procedure Development Report
The outcome of Stage 1 should be a comprehensive method development report describing the optimized procedure. It should also contain practical details for the procedure, including the robustness of the analytical procedure, the analytical control strategy and the SST parameters to be used, and their acceptance criteria.
This is in stark contrast with the FDA bioanalytical method and draft ICH M10 guidance documents that suggest that bioanalytical method development does not require extensive recordkeeping or notation (10,11). If you don’t have any understanding of how critical parameters impact the performance of an analytical procedure, then how can you control them? In my opinion, method development needs a report that highlights those key parameters, and how they impact performance of the procedure. It is good analytical science, and an essential reference for all further work.
Stage 2: Procedure Performance Qualification
Planning the Validation
Procedure Performance Qualification (PPQ) or method validation should be simply confirmation of good method development and demonstrate that the analytical procedure is fit for purpose. PPQ demonstrates that the developed analytical procedure meets the ATP quality attributes, and that the performance is appropriate for the intended use. To control the work, there will be a validation plan or protocol describing the experiments to be performed, with predefined acceptance criteria to demonstrate that the ATP has been met. This will depend on the type of procedure, such as active pharmaceutical ingredient (API), impurities, or bioanalysis. The various experiments will depend on the criteria described in the ATP, and on the intended use of the procedure.
For example:
- Linearity experiments should be used to support the use of the specific calibration model used in the procedure (the calculations for which have been verified in Level 1 in the computerized system validation of the data system used for this work).
- Specificity or selectivity (depending on whether the instrumental technique is absolute or comparative) must be determined, including resolution for impurities and peak purity assessment for stability-indicating methods.
- Precision (injection precision, repeatability, and intermediate precision) should be set. The minimum number of runs could be two, but four or more provides better understanding of the intermediate precision for routine use
- Accuracy can be run in the same experiments as precision.
- Analyte stability under storage, laboratory, and instrument conditions must be determined.
- System suitability test parameters, and their acceptance criteria, will be verified during this work
It is important that the acceptance criteria be defined in the validation plan, and are based on the information gathered from Stage 1, the procedure design and development. The plan will also define how the data from the various experiments will be evaluated statistically against the acceptance criteria.
Validation Report
Once the work is completed, a report is written that describes the outcome of the validation experiments and how the procedure meets the requirements of the ATP.. As the draft USP <1220> (12) notes:
The analytical control strategy may be refined and updated as a consequence of any learning from the qualification study. For example, further controls may be added to reduce sources of variability that are identified in the routine operating environment in an analytical laboratory, or replication levels (multiple preparations, multiple injections, etc.) may be modified based on the uncertainty in the reportable value.
The scope and the various parameters with the acceptance criteria for a bioanalytical method validation report are defined extensively in the updated FDA Guidance for Industry on Bioanalytical Method Validation and the draft ICH M10 guidance documents (10,11).
Analytical Procedure Transfer
Analytical method transfer is not always easy or straightforward, because there are always items that are not well described in, or even omitted altogether from, an analytical procedure. Well-documented method development (if existing) and validation reports will aid the transfer process immeasurably. The transfer must be planned, and a protocol developed, between the originating and receiving laboratories that includes predefined ways that the data will be interpreted with acceptance criteria. A report should be produced summarizing the transfer results against the data generated by the receiving laboratory.
To reduce the effort required when transferring an analytical procedure to another laboratory, a subject-matter expert could travel to the receiving laboratory to provide help and advice. Alternatively, an analyst from the receiving laboratory could go to the originating laboratory to learn the procedure. Management often looks at the up-front cost of this, but dismisses the hidden cost of time wasted in transferring the method without help from the originating laboratory.
When considering method transfer, one of the issues when using a contract research organization (CRO) laboratory is the quality of the written procedure used for method transfer. Often the originating laboratory (sponsor) may make a minimal effort at validation before passing the procedure to a CRO to complete the development and validation. This is not the best approach, and is planning for failure.
Stage 3: Procedure Performance Verification
Routine monitoring of an analytical procedure’s ongoing performance is an important element in maintaining control over the analytical procedure in operational use. It provides assurance that the analytical procedure remains in a state of control throughout its lifecycle, and provides a proactive assessment of a procedure’s performance. The aim of verification is that the reportable result is fit for purpose and can be used to make a decision.
Part of this verification can be trending of SST and sample replicates results over time. However, there is a note of caution that SST results can also be used to measure instrument performance directly (Group B and some Group C instruments) or indirectly (some Group C instruments) as part of an ongoing performance qualification. Data that could be collected and tracked are:
- SST test results including failures
- Trending of individual results and the reportable result including OOS and outputs from investigations.
These data should be monitored against limits, so that when there is a trend indicating a parameter is out of control, an investigation can be started early, before the situation gets out of hand (a proactive, rather than reactive, approach). When a root cause is identified in an investigation, it may be appropriate to update the analytical control strategy or to update the analytical procedure.
Pharma Is Going Lifecycle
Why have I discussed this new approach? The pharmaceutical industry is going lifecycle! ICH published in November 2019 a new guidance, ICH Q12, entitled “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management” (16). This document provides a framework to facilitate the management of post-approval Chemistry Manufacturing Controls (CMC) changes in a more predictable and efficient manner and the key concept is Established Conditions (ECs) for analytical procedures:
ECs related to analytical procedures should include elements which assure performance of the procedure. The extent of ECs and their reporting categories could vary based on the degree of the understanding of the relationship between method parameters and method performance, the method complexity, and control strategy (16)
Note that ECs are legally binding, as they will be part of a drug license application and changes will need regulatory approval. ECs are built up during the development and validation process of the lifecycle.
In addition, to further support the analytical procedure lifecycle management (APLM) approach, ICH is undertaking two projects:
- An update and expansion of ICH Q2(R1) to include other analytical techniques with a possible release for public comment of Q2(R2) as early as the end of this year (17)
- ICH Q14 on Analytical Procedure Development, which is beginning to be developed (18).
Common sense would suggest that combining the two into a single document would be the best approach. However, putting common sense and regulatory compliance in the same breath would be a novel idea.
What this means in practice is that the more you know about your analytical procedure, the more predictable the analysis becomes, thanks to the lower variation. There should be a lower regulatory burden to change a registered method. Most importantly, with robust analytical procedures there should be a lower incidence of OOS results attributed to analytical variation and subsequent investigations. OOS will be the subject of an article later in our series.
Summary
A key component for data integrity is accurate and precise analytical procedures that are validated for intended use. Changes in the way procedures are specified, developed, validated, and operated are coming. This “Data Integrity Focus” article should help prepare you for the changes.
Acknowledgments
I would like to thank Chris Burgess for review of this article.
References
- R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
- R.D. McDowall, LCGC N. Am.37(1), 44–51 (2019).
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Sliver Spring, Maryland, 2008).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control (European Commission, Brussels, Belgium, 2014).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15 Qualification and Validation (European Commission, Brussels, Belgium, 2015).
- ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology (International Conference on Harmonisation, Geneva, Switzerland, 2005).
- FDA Draft Guidance for Industry: Analytical Procedures and Methods Validation (Food and Drug Administration, Rockville, Maryland, 2000).
- FDA Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics (Food and Drug Administration, Rockville, Maryland, 2015).
- EMA Guideline on Bioanalytical Method Validation (European Medicines Agency, London, United Kingdom, 2011).
- FDA Guidance for Industry: Bioanalytical Methods Validation (Food and Drug Administration, Rockville, Maryland, 2018).
- ICH M10 Bioanalytical Method Validation Stage 2 Draft (International Council on Harmonisation, Geneva, Switzerland, 2019).
- G.P. Martin, et al., Pharmacopoeial Forum 43(1), (2017).
- J. Ermer and P. Nethercote, Method Validation in Pharmaceutical Analysis, A Guide to Best Practice (Wiley-VCH,Weinheim, Germany, 2nd Ed., 2015).
- E. Kovacs, et al., Pharmacopoeial Forum 42(5), (2016).
- K.L. Barnett et al., Pharmacopoeial Forum 42(5), (2016).
- CH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management (International Council for Harmonisation Geneva, Switzerland, 2019).
- Concept Paper: Analytical Procedure Development and Revision of ICH Q2(R1) Analytical Validation (International Council on Harmonisation: Geneva, Switzerland, 2018).
- Final Concept Paper ICH Q14: Analytical Procedure Development and Revision of Q2(R1) Analytical Validation (International Council on Harmonisation: Geneva, Switzerland, 2018).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com















Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



