Trends in Separation Science: Tackling Protein Analysis
Leading separation scientists discuss their approaches to improving the analysis of large molecules.
Increasingly, separation scientists are turning their attention to the challenges of analyzing proteins, whether in pharmaceutical development or proteomics. Heading into the HPLC 2016 conference in San Francisco this June, we asked several leading analytical chemists, all of whom who will be speaking at the conference, to talk about their work in this area. Below, John Yates of the Scripps Research Institute, Mary Wirth of Purdue University, Andrew Alpert of PolyLC, and Alain Beck of the Pierre Fabre Immunology Center and Arnaud Delobel of Quality Assistance SA discuss approaches to improve the analysis of these large molecules.
Why Effective HPLC Separations Are Critical for the Analysis of High-Complexity Systems
John Yates III
Large-scale protein biochemistry, otherwise known as proteomics, was enabled by the protein sequence infrastructure created by genome sequencing. Tandem mass spectrometers create spectra of peptides that can be used to search sequence databases to assign amino acid sequences to the spectra. Such methods allow the analysis of protein complexes, organelles, and whole cells through a process that proteolytically digests proteins into a collection of peptides that can then be identified using tandem mass spectrometry (MS) and informatics. Intrinsic to this strategy is the creation of very large and complicated mixtures of peptides. Simultaneous electrospray ionization of large and diverse collections of peptides results in suppression of ion formation; consequently it is imperative to separate peptides sufficiently to relieve ion suppression and to allow the mass spectrometer sufficient time to collect tandem mass spectra of peptide ions. Thus, peptide separation methods are critical for proteomics studies. The challenges involved are great.
The digestion of the proteins from a human cell can produce many hundreds of thousands of peptides. The analysis of such a complex mixture by tandem MS is dependent on both the resolution of the separation and also on the scan speed of the tandem mass spectrometer. Separation resolution has to decrease the complexity of the mixture so the instrument can maximize collection of tandem mass spectra of peptide ions. The scan speeds of tandem mass spectrometers are increasing, allowing the collection of more spectra per unit time. New data acquisition strategies like data-independent data acquisition are threatening to further increase data acquisition rates. Clearly, this situation creates a challenging separation problem, especially if the goal is to also minimize the overall time required for the analysis.
Initial methods to resolve such complicated peptide mixtures used a two-dimensional liquid chromatography (2D LC) separation technique that combined a strong cation exchange separation with reversed-phase separation using a C18 column (1,2). While not perfectly orthogonal, the two modes provide excellent peak capacity, but do so with long analysis times to achieve high numbers of protein identifications. Various other 2D approaches to this problem have been developed, including a high-pH reversed-phase separation followed by a low-pH reversed-phase separation and methods that combine a strong anion exchange step with a reversed-phase step, analogous to the strong cation exchange-reversed phase combination (3).
Through continuous improvements to MS technology, it is now possible to analyze most of a mammalian cell proteome. In fact, a nearly complete analysis of the eukaryotic Saccharomyces cerevisiae proteome has been reported (4) with an analysis time under 90 min using reversed-phase high-performance LC (HPLC). By decreasing analysis times, higher throughput experiments can be designed that can examine more human patient samples or more sample conditions. The development of high-resolution single-dimension separations based on ultrahigh-pressure liquid chromatography (UHPLC) using a C18 column has offered a new direction, although the time required to achieve high numbers of peptide identifications is still 4â8 h for complex proteomes (5,6). Additionally, those analyses achieve nearly complete protein analyses but with very low numbers of identified peptides per proteins (in other words, low sequence coverage) and in fact most of the proteome is identified with one peptide per protein.
Another reason to increase the resolution of separations and peak capacities is to improve the dynamic range of the separation. Within a proteome there is at least an abundance range of 106 for expressed proteins and this range doesn’t include post-translational modifications (PTMs). It remains to be seen if single-dimension UHPLC separations can be extended to encompass the entire proteome of a mammalian cell to capture all the subtleties and nuances of protein structure. Developing high-speed 2D LC separations may provide a means to achieve this goal, but this type of method does present a challenge because of the slower analysis times often associated with separations in two dimensions.
As separation methods in combination with advanced MS technology bring us closer to the capability to characterize the complete human proteome, advancing these methods to achieve high sequence coverage will be essential. High sequence coverage in combination with good dynamic range will allow collection of information about isoforms and PTMs, information, which will be important to understand the function of proteins.
References
- A.J. Link et al., Nat. Biotechnol. 17, 676â682 (1999).
- M.P. Washburn, D. Wolters, and J.R. Yates, Nat. Biotechnol. 19, 242â247 (2001).
- M. Gilar, P. Olivova, A.E. Daly, and J.C. Gebler, Anal. Chem.77, 6426â6434 (2005).
- A.S. Hebert et al., Mol. Cell Proteomics13, 339â347 (2014).
- Q. Luo et al., Anal. Chem.77, 5028â5035 (2005).
- J.E. MacNair, K.D. Patel, and J.W. Jorgenson, Anal. Chem.71, 700â708 (1999).
John Yates III is the Ernest W. Hahn Professor of Chemical Physiology and Molecular and Cellular Neurobiology at The Scripps Research Institute, in La Jolla, California.
New Materials for UHPLC Analysis of Monoclonal Antibody Drugs and AntibodyâDrug Conjugates
Alexis Huckabee, Jonathan Yasosky, and Mary J. Wirth
The separation of intact proteins plays many roles in drug discovery and development: identification of drug targets, screening for drug candidates, quality control, and drug authentication. Various separation techniques are used, from immunoprecipitation to study a single protein of interest, through various types of column chromatography for detecting a handful of proteins at once, all the way to proteomics for studying hundreds to thousands of proteins. What all of these techniques and applications have in common is that the power of protein separations is limited by the fact that proteins are large, slowly diffusing, sticky molecules.
There is a demand for even higher performance. One critical need for better chromatography is in the development of antibodyâdrug conjugates (ADCs). Only a few ADCs have been approved by the US Food and Drug Administration (FDA), but many ADCs are under development and in all phases of clinical trials. An ADC directs a toxic small-molecule drug to cancer cells by covalently binding the toxic drug to a monoclonal antibody through a labile bond (1). The intention is that the labile bond breaks after the ADC is inside the cancer cell, but not before, to release the toxin where it is needed. ADCs have multiple binding sites, and each species potentially has different efficacy and toxicology. Therefore, it is necessary to determine to how many of these binding sites the drug has been attached. Hydrophobic interaction chromatography (HIC) uses columns packed with nonpolar polymer beads to separate ADCs based on how many drug molecules are conjugated to a given antibody (the drugâantibody ratio, or DAR) (2). The bands typically are too broad to fully separate all of the species, however, and some ADC candidates are too hydrophobic for current HIC columns. Decades of research and development (R&D) in the chromatography industry have been devoted to reversed-phase liquid chromatography (LC) because this mode of LC is so widely used for small-molecule drugs and for quality control of protein drugs. Creating better bonded phases for HIC, however, is a challenge for silica-based columns because HIC uses a fully aqueous mobile phase and neutral pH.
The bonded phase is a pervasive limitation to efficiency in monoclonal antibody separations. Another problem with monoclonal antibody separations is that silica particles having an average pore diameter of 300 Ã do not allow full access to the surface area. The limited pore size also impedes intraparticle diffusion. Advances in the chromatographic materials need to consider all of these issues.
Bonded Phases with Less Protein Adhesion
There are two approaches to reducing silanol activity in HIC columns that our group has considered. One is self-assembled monolayers for reversed-phase separations, illustrated in Figure 1a. These monolayers form a dense, two-dimensional net over the surface to improve reversed-phase LC separations of proteins (3). Since coulombic interactions are long-range, any defects in the column produce the familiar tailing of mixed-mode chromatography techniques, which is mitigated by low pH. The other approach, surface-confined atom-transfer radical polymerization, alleviates the coulombic interactions by growing a dense layer of tethered polymer chains that is thicker than the Debye length, as illustrated in Figure 1b, reducing long-range attractions between proteins and silica (4). Nonporous polymer particles with tethered polymers as bonded phases (5) have been commercially available for some time. Despite their small surface area, these particles have been valuable for monoclonal antibody separations, including ion-exchange chromatography and HIC, as mentioned earlier. Our group found that the bonded phase depicted in Figure 1b gives excellent hydrophilic-interaction chromatography (HILIC), resolving the glycosylation states of ribonuclease B and providing partial resolution of their isomers (6). We are currently applying HILIC to monoclonal antibodies to decrease analysis time for probing glycosylation. We are also investigating whether the combination of self-assembly and tethered polymer chains on silica can result in a better HIC bonded phase for ADC analysis.


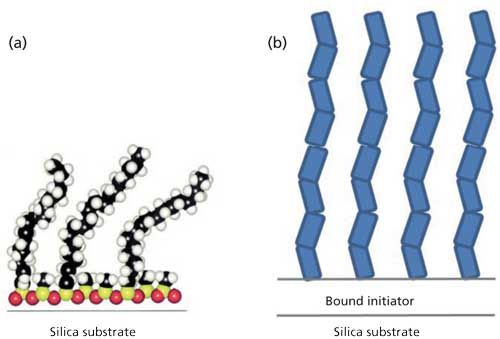
Figure 1: Two bonded phases that reduce silanol interactions: (a) self-assembled mixed silanes and (b) densely grown polymer chains tethered from silica. These images are not to scale; the polymer layer is typically about 10 times thicker than the C18 layer.
Morphologies to Decrease Diffusion Distance and Increase Accessible Surface Area
The most salient commercial advance in LC over the last 15 years has been the introduction of chromatographic particles that decrease the diffusion distances for mass transport. These include fully porous sub-2-μm particles (7), coreâshell particles (8), and monolithic columns (9). Porous materials, regardless of morphology, will eventually approach the limit where the smaller particle size (or smaller shell thickness) approaches the diameter of the larger colloids needed to produce wider pore sizes (10). Sphere-on-sphere particles, which are coreâshell particles with a single layer of large colloids (11), embody this idea. As this limit is approached for fully porous particles, it makes sense to consider using the colloids themselves as the packing material.
We have studied capillaries packed with 0.5-μm nonporous silica particles with C4 bonded phases, packed in capillaries. The accessible surface area for large proteins is quite high, falling in between those of coreâshell particles and fully porous particles, each for a 300-à pore size (12). The back pressure is alleviated by the presence of slip flow for reversed-phase LC (13), enabling column lengths of at least 10 cm for commercial high-pressure chromatography instruments. Colloidal particles pack more tightly than standard particles, obstructing the axial diffusion about twofold more. This tighter packing gives rise to a higher velocity inside the column, which makes the optimal volume flow rate slower, further alleviating back pressure. Uniform packing results in negligible eddy dispersion. With no intraparticle diffusion, lower obstructed diffusion, and lower eddy dispersion, all three terms in the van Deemter equation are thus smaller. Barring effects from strong adsorption of proteins, the van Deemter equation predicts submicrometer plate height for proteins.
We observed submicrometer plate heights for protein separations using 0.5-μm particles (12,13,15). Plate heights were as small as 15 nm and the plate number was in excess of 106 for a 2-min separation of proteins (13). In this case, self-assembly was used to produce a mixed C4âC1 bonded phase, injection was done by diffusing the protein into the head of the column to avoid contact between metal parts and protein, and on-column detection was used. For more-routine applications, Figure 2 shows an example separation where a commercial auto-injector was used, with the same bonded phase and on-column detection. An IgG4 monoclonal antibody drug and its two aggregates were resolved well in a short 1-cm separation length at room temperature, whereas a 5-cm long column of fully porous particles did not resolve the three species.

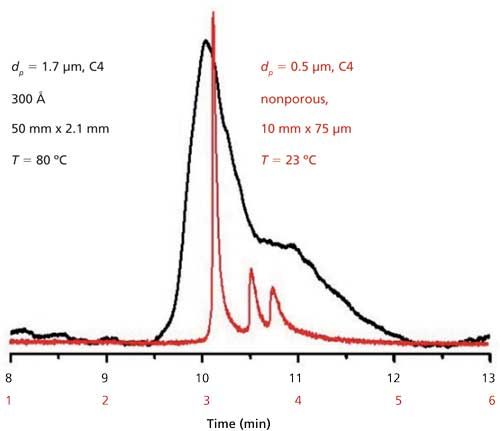
Figure 2: Reversed-phase LC data for a monoclonal antibody and aggregates for a commercial column of fully porous sub-2-μm particles (black) and a capillary packed with 0.5-μm particles (red), each with a C4 bonded phase. Adapted with permission from reference 14.
HIC columns will likely enjoy a boost in flow rate from slip flow when used with submicrometer particles. The next goals are to extend the performance seen in Figure 2 to 2.1-mm i.d. stainless steel columns for reversed-phase LC, and combine this advance with a new polymeric bonded phase on silica for HIC of ADCs.
References
- P. Polakis, Pharmacol. Rev. 68, 3â19 (2016).
- M. Rodriguez-Aller, D. Guillarme, A. Beck, and S. Fekete, J. Pharm. Biomed. Anal. 118, 393â403 (2016).
- M.J. Wirth, R.W.P. Fairbank, and H.O. Fatunmbi, Science 275, 44â47 (1997).
- X.Y. Huang and M.J. Wirth, Anal. Chem. 69, 4577â4580 (1997).
- M. Weitzhandler, D. Farnan, J. Horvath, J.S. Rohrer, R.W. Slingsby, N. Avdalovic, and C. Pohl, J. Chromatogr. A828, 365â372 (1998).
- Z. Zhang, Z. Wu, and M.J. Wirth, J. Chromatogr. A1301, 156â161 (2013).
- J.E. MacNair, K.C. Lewis, and J.W. Jorgenson, Anal. Chem. 69, 983â989 (1997).
- J.J. Kirkland, Anal. Chem. 64, 1239â1245 (1992).
- N. Ishizuka, H. Minakuchi, K. Nakanishi, N. Soga, and N. Tanaka, J. Chromatogr. A797, 133â137 (1998).
- X.Y. Huang, R.S. McLean, and M. Zheng, Anal. Chem.77, 6225â6228 (2005).
- A. Ahmed, H. Ritchie, P. Myers, and H. Zhang, Adv. Mater.24, 6042 (2012).
- B.J. Rogers and M.J. Wirth, J. Phys. Chem. A117, 6244â6249 (2013).
- B. Wei, B.J. Rogers, and M.J. Wirth, J. Am. Chem. Soc.134, 10780â10782 (2012).
- B.J. Rogers, R.E. Birdsall, Z. Wu, and M.J. Wirth, Anal. Chem. 85, 6820â6825 (2013).
- B.A. Rogers, Z. Wu, B.C. Wei, X.M. Zhang, X. Cao, O. Alabi, and M.J. Wirth, Anal. Chem.87, 2520â2526 (2015).
Mary J. Wirth is the W. Brooks Fortune Professor of Chemistry at Purdue University in West Lafayette, Indiana. Alexis Huckabee and Jonathan Yasosky are graduate research assistants in the department.
Reconsidering HIC for Top-Down Proteomics
Andrew J. Alpert
Until recently, mass spectrometry (MS) was limited in the information it could supply regarding proteins larger than 40 kDa. The most recent instruments have broken through that limit, but proteins smaller than 40 kDa are still more easily detected in MS and can suppress the collection of data from larger proteins. This situation has created a demand for better separation of proteins upstream from the MS orifice to facilitate top-down proteomics. At present, though, this separation of proteins is something of a bottleneck. Methods such as reversed-phase liquid chromatography (LC) that involve mobile phases compatible with MS are not compatible with many proteins. With those mobile- and stationary-phase combinations, the proteins may be eluted in peaks 15-min wide or not at all (1,2). The conditions also tend to cause the loss of protein three-dimensional structure, precluding analysis of protein complexes.

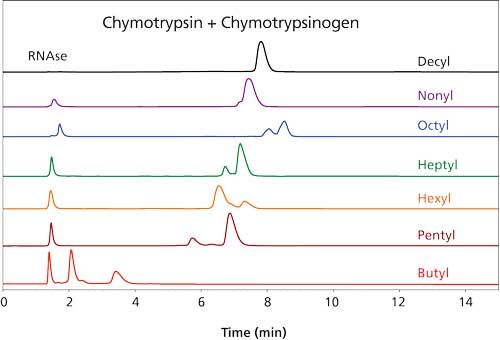
Figure 1: Retention of ribonuclease A (RNAse) and chymotrypsinogen (CHYGEN) on various columns. Columns: 100 mm x 4.6 mm, 3-µm dp, 1500-à (coating as shown); mobile-phase A: 1 M ammonium acetate; mobile-phase B: 20 mM ammonium acetate with 50% acetonitrile; gradient: 15-min linear, 0â100% B; flow rate: 1 mL/min; detection: 280 nm. (Adapted with permission from reference 6.)
Alternative modes of chromatography for protein analysis include the following:
- Size-exclusion chromatography: Size-exclusion chromatography is helpful in separating large proteins from small ones but is otherwise a low-resolution method.
- Ion-exchange chromatography: Ion-exchange chromatography is a high-resolution mode but generally requires more salt than a mass spectrometer can cope with.
- Hydrophilic-interaction chromatography (HILIC): HILIC has been used successfully for proteins that do not normally occur free in aqueous solution, such as histones (3), membrane proteins (4), and apolipoproteins (5). However, the high concentration of organic solvent required probably precludes its more general use.
- Hydrophobic interaction chromatography (HIC): HIC involves a gradient from a high to low concentration of salt, and the best-performing salts are nonvolatile. This condition would seem to eliminate it from consideration for protein separations online with MS. However, HIC separates proteins with high resolution, based on differences in the hydrophobicity of the surface of their tertiary structures. HIC is nondenaturing and extremely sensitive to differences in protein composition. Accordingly, we decided to take another look at HIC (6).
The use of HIC would be practical if a volatile salt could be used. Suitable salts such as ammonium acetate are poor at promoting retention in the HIC mode. Literature on the subject has involved concentrations in the 3â4 M range, which is too high for a mass spectrometer. Now, the more hydrophobic a material is, the less salt is needed for retention in HIC. We decided to increase the hydrophobicity of the stationary phase systematically in an effort to produce materials that could retain proteins at concentrations of ammonium acetate that were practical for MS analysis. Increasing the length of the functional ligand in the coating from C3 to C4 to C5 resulted in dramatic increases in protein retention, in keeping with past observations about HIC (7). However, lengthening the ligand from C5 through C10 did not result in much change in protein retention times (Figure 1). Furthermore, in this range the retention of some of the protein standards was not directly proportional to the concentration of ammonium acetate (Figure 2). This behavior is normally associated with reversed-phase LC rather than with HIC. A concentration of 0.75â1 M ammonium acetate is still necessary in the starting mobile phase, but its function seems to be the maintenance of the tertiary structure of the proteins rather than promotion of binding. Mass spectrometers can handle such concentrations. The presence of some organic solvent in the final mobile phase was essential for elution of most proteins within the gradient. All in all, the behavior of these new HIC materials resembles that of reversed-phase LC as much as HIC. The distinction between the two modes seems to have been blurred if not erased.

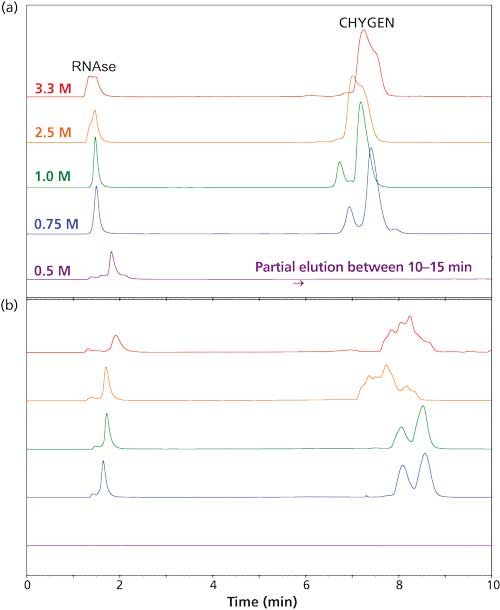
Figure 2: Retention of RNAse and CHYGEN as a function of initial salt concentration: (a) PolyHEPTYL A column, (b) PolyOCTYL A column. Mobile-phase A: ammonium acetate concentration as noted; mobile-phase B: 20 mM ammonium acetate with 50% acetonitrile; gradient: 15 min, 0â100% B, then 5 min at 100% B. Other running conditions were the same as in Figure 1. (Adapted with permission from reference 6.)
Figure 3 demonstrates that standard proteins are eluted from the new materials with good peak shape. Direct elution into a mass spectrometer produced mass spectra characteristic of proteins with their native structures intact. Notwithstanding the high concentration of acetonitrile in the final mobile phase, the kinetics of the chromatography was evidently faster than the kinetics of denaturation. Figure 4 is a demonstration of direct HICâMS of an extract of E. coli proteins. A protein as large as 206 kDa was identified.

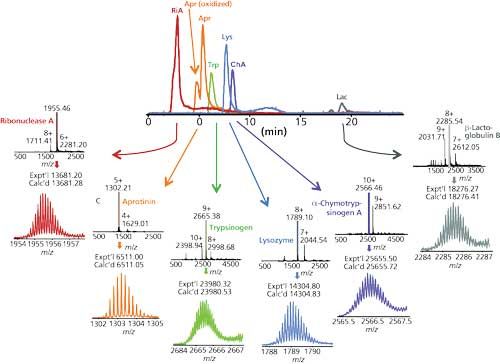
Figure 3: HIC separation of standard proteins and Q-TOF-MS analysis. Column: 100 mm x 0.2 mm, 3-µm dp, 1500-à PolyHEPTYL A capillary; mobile-phase A: 1 M ammonium acetate; mobile-phase B: 20 mM ammonium acetate with 50% acetonitrile; gradient: 15-min linear, 0â100% B, then 5 min at 100% B; flow rate: 2.4 µL/min; detection: Q-TOF-MS. (Adapted with permission from reference 6.)

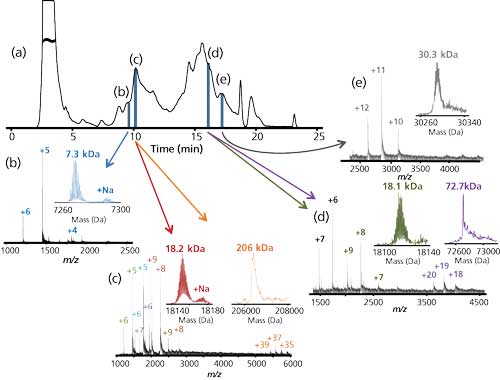
Figure 4: HIC separation of standard proteins and Q-TOF-MS analysis. Column: 100 mm x 0.2 mm, 3-µm dp, 1500-à PolyHEPTYL A capillary; mobile-phase A: 1 M ammonium acetate; mobile-phase B: 20 mM ammonium acetate with 50% acetonitrile; gradient: 15-min linear, 0â100% B, then 5 min at 100% B; flow rate: 2.4 µL/min; detection: Q-TOF-MS. (Adapted with permission from reference 6.)
This project seems to have been successful in adapting HIC to the requirements of MS for top-down proteomics. It should be possible to adapt ion-exchange chromatography for this purpose as well.
Acknowledgments
This work was performed in collaboration with Ying Ge of the University of Wisconsin-Madison, and the following members of her group: Bifan Chen, Ying Peng, Santosh G. Valeja, and Lichen Xiu.
References
- A. Vellaichamy, J.C. Tran, A.D. Catherman, J.E. Lee, J.F. Kellie, S.M.M. Sweet, L. Zamdborg, P.M. Thomas, D.R. Ahlf, K.R. Durbin, G.A. Valaskovic, and N.L Kelleher, Anal. Chem.82, 1234â1244 (2010).
- M.B. Strader, N.C. VerBerkmoes, D.L. Tabb, H.M. Connelly, J.W. Barton, B.D. Bruce, D.A. Pelletier, B.H. Davison, R.L. Hettich, F.W. Larimer, and G.B. Hurst, J. Proteome Res. 3, 965â978 (2004).
- N.L. Young, P.A. DiMaggio, M.D. Plazas-Mayorca, R.C. Baliban, C.A. Floudas, and B.A. Garcia, Mol. Cell. Proteomics8, 2266â2284 (2009).
- J. Carroll, I.M. Fearnley, and J.E. Walker, Proc. Natl. Acad. Sci. USA103, 16170â16175 (2006).
- T. Tetaz, S. Detzner, A. Friedlein, B. Molitor, and J.-L Mary, J. Chromatogr. A1218, 5892â5896 (2011).
- B. Chen, Y. Peng, S.G. Valeja, L. Xiu, A.J. Alpert, and Y. Ge, Anal. Chem. 88, 1885â1891 (2016).
- A.J Alpert, J. Chromatogr.359, 85â97 (1986).
Andrew J. Alpert is the president of PolyLC Inc. in Columbia, Maryland.
State-of-the-Art MS Methods for Structural Assessment of mAbs and ADCs: From the Research Lab to Routine Characterization
Alain Beck and Arnaud Delobel
Monoclonal antibodies (mAbs) are highly complex tetrameric glycoproteins that require extensive analytical and structural characterization to become drug candidates. This is also true for antibodyâdrug conjugates (ADCs). These immunoconjugates are based on highly cytotoxic small-molecule drugs covalently attached via conditionally stable linkers to mAbs and are among the most promising next-generation empowered biologics for cancer treatment. ADCs are more complex than naked mAbs, because the heterogeneity of the conjugates adds to the inherent microvariability of the biomolecules. The development and optimization of ADCs rely on improving their analytical and bioanalytical characterization by assessing several critical quality attributes, namely the distribution and position of the drug, the amount of naked antibody, the average drug-to-antibody ratio (DAR), and the proportions of residual small-molecule drug and drug-linker (1).
As a result of advances in multilevel (top, middle, bottom) state-of-the-art mass spectrometry (MS) methods, including native MS, ion mobility MS (2), capillary electrophoresisâelectrospray ionizationâMS (3,4), two-dimensional liquid chromatographyâMS (2D LCâMS) (5), extended bottom-up (6), and top-down sequencing (7), combined with chromatographic and electrophoretic techniques (8) very precise characterization of biotherapeutics is now possible. Until recently, however, these techniques were considered suitable only for research use. With the advent of robust and user-friendly solutions (both hardware and software), these techniques are now amenable for routine use. Examples of their application to the characterization of mAbs and ADCs are discussed below.
Middle-Level Characterization of mAbs
During their biosynthesis or during their shelf life, mAbs can undergo many modifications, such as glycosylation, oxidation, deamidation, and C-terminal lysine clipping, to name a few. In most cases, these variants cannot be easily identified at the intact antibody level, because of limitations in chromatographic separation and MS resolution.
A middle-up approach using IdeS digestion (to yield Fab, Fc/2, and light-chain fragments) combined with a super macroporous reversed-phase column enables quick and efficient characterization of mAb variants (9). The relatively low molecular mass of the subunits (~25 kDa) allows accurate mass determination by high-resolution MS as well as top-down sequencing by electron transfer dissociation (ETD). Using the same sample preparation, the glycoforms can also be separated and characterized using an approach such as hydrophilic-interaction chromatography (HILIC) with MS detection (10). An example of this approach is presented in Figure 1 for adalimumab.

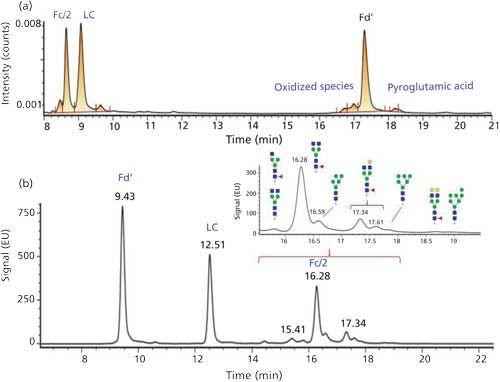
Figure 1: HPLCâUV chromatograms obtained for adalimumab sample digested with IdeS and analyzed on (a) a reversed-phase or (b) a HILIC column.
Determination of Drug-to-Antibody Ratio on Intact ADCs
The number of cytotoxic molecules attached to an antibody (the drug-to-antibody ratio, or DAR) is a critical quality attribute of an ADC. It can be determined by UV spectrophotometry or hydrophobic interaction chromatography (HIC) with UV detection (11â13), but these methods are not universal and have some drawbacks. Mass spectrometry can be a universal tool to determine the DAR value, whatever the coupling chemistry or the nature of the drugs.
After N-deglycosylation, the ADC is desalted on-line using reversed-phase high performance LC (HPLC) or size-exclusion chromatography (SEC) (14) and detected by high-resolution electrospray ionization MS. The DAR can be automatically calculated by software after deconvolution of the multicharged electrospray spectrum. When working with cysteine-linked ADCs, native conditions must be used to avoid disruption of noncovalent interactions. In these methods, native SECâMS is commonly used, which requires optimization of mobile phases and MS conditions.
Characterization of mAbs and ADCs by UHPLCâMS and HPLCâMS Peptide Mapping
Peptide mapping with MS detection is a common methodology for protein characterization. It can be used for the confirmation of the primary sequence, the quantification of post-translational modifications (PTMs), and the study of disulfide-bond scrambling. Thanks to commercially available software packages dedicated to biopharmaceutical analysis, the analysis of these MS data can be fully automated.
Applied to ADC characterization, peptide mapping is also a valuable tool to localize conjugation sites and determine site occupancy. ETD fragmentation can be used to localize conjugation sites for lysine-conjugated ADCs on peptides containing several lysine residues.
2D LCâMS Analysis of mAbs and ADCs
Two-dimensional LC with MS detection is widely used for the identification of proteins from complex proteome samples in many laboratories. Robust 2D-HPLC and 2D-ultrahigh pressure LC (UHPLC) systems are now commercially available, thus enabling the routine analysis of biopharmaceuticals with this technology.
Two-dimensional LC can be used to hyphenate MS-incompatible chromatographic separations to mass spectrometry detection: After a first dimension using mobile phases containing nonvolatile salts, the peak of interest is sent to a second dimension consisting of a reversed-phase column to desalt the sample and separate potentially coeluted species. This methodology (heart-cutting 2D LCâMS) can be routinely applied to identify the different species detected in size-exclusion and ion-exchange chromatography of monoclonal antibodies.
Another main application of 2D LCâMS is the rapid on-line structural elucidation of species observed in HIC distribution profiles of cysteine-conjugated ADCs (15,16). The identification of the different species is required to be able to determine the DAR value based on the HICâUV profile as well as for the detection and the quantification of small-molecule drugs (17). The application of this methodology to brentuximab vedotin is presented in Figure 2.

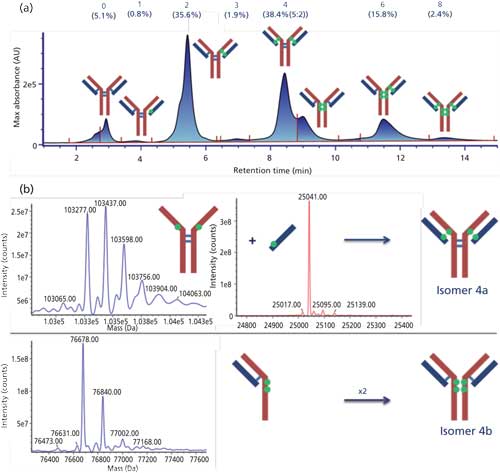
Figure 2: (a) HICâUV chromatogram obtained for brentuximab vedotin and (b) deconvoluted mass spectra obtained by HICâreversed-phase LCâMS for the two isomers of DAR4.
References
- A. Beck, G. Terral, F. Debaene, E. Wagner-Rousset, J. Marcoux, MC Janin-Bussat, O. Colas, A. Van Dorsselaer, and S. Cianferani, Exp. Rev. Proteomics13(2), 157â183 (2016).
- G. Terral, A. Beck, and S. Cianferani, J. Chromatogr. B, in press.
- R. Gahoual, A. Beck, Y.N. François, and E. Leize-Wagner, J. Mass Spectrom. 51(2), 150â158 (2016).
- Y.N. François, M. Biacchi, N.Said, C. Renard, A. Beck, R. Gahoual, and E. Leize-Wagner, Anal. Chim. Acta908, 168â176 (2016).
- D. Stoll, J. Danforth, K. Zhang, and A. Beck, J. Chromatogr. B, in press.>
- N. Gasilova, K. SrzentiÄ, L. Qiao, B. Liu, A. Beck, Y.O. Tsybin, and H.H. Girault, Anal. Chem.88, 1775â1784 (2016).
- R Gahoual, A. Beck, E. Leize-Wagner, and Y.N. François, J. Chromatogr. B, in press.
- A. Resenman, W. Jabs, A. Wiechmann, E Wagner-Rouset, O. Colas, W. Evers, E. Belau, L. Vorwerg, C. Evans, A. Beck, and D. Suckau, mAbs8(2), 318â330 (2016).
- S. Sjögren, F. Olsson, and A. Beck, Analyst, in press.
- A. Peria, S. Fekete, A. Cusumano, J.L. Veuthey, A. Beck, M. Lauber, and D. Guillarme, J. Chromatogr. A, in press.
- M. Rodriguez-Aller, D. Guillarme, A. Beck, and S. Fekete, J. Pharm. Biomed. Bioanal.118, 393â403 (2016).
- A. Cusumano, D. Guillarme, A. Beck, and S. Fekete, J. Pharm. Biomed. Bioanal. 121, 161â173 (2016).
- S Fekete, J.L. Veuthey, A Beck, and D. Guillarme, J. Chromatogr. B, in press.
- M. Rodriguez-Aller, A. Cusumano, A Beck, D. Guillarme, and S. Fekete, J Chromatogr. B, in press.
- M. Sarrut, A. Corgier, S. Fekete, D. Guillarme, D. Lascoux, M.C. Janin-Bussat, A. Beck, and H. Heinisch, J. Chromatogr. B, in press.
- M. Sarrut, M.C. Janin-Bussat, S. Fekete, D. Guillarme, A. Beck, and H. Heinisch, J. Chromatogr. B, in press.
- R.E. Birdsall, S.M. McCarthy, M.C. Janin-Bussat, M. Perez, J.F. Haeuw, C. Weibin, and A. Beck, mAbs8(2), 306â317 (2016).
Alain Beck is the Senior Director of the Physico-Chemistry Department at the Centre d’Immunologie Pierre Fabre (CIPF) in Saint-Julien-en-Genevois, France. Arnaud Delobel is the scientific manager for biologics and the R&D team manager at Quality Assistance SA, in Donstiennes, Belgium.
Direct correspondence about this article to: LCGCedit@ubm.com.


















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.



