Too Much Scrutiny
Look at the big picture. It may not be appropriate to obsess on the details
As I write this column, I have just returned from the Pittsburgh Conference in Orlando, Florida. One of the things that I enjoy about Pittcon is doing booth duty, when I get to meet some of you loyal readers of "LC Troubleshooting". Sometimes the conversations turn to specific problems that you are having with your liquid chromatography (LC) system or separation. Often these can be a mind-stretching conversation with give-and-take that is not as convenient in an e-mail exchange. However, some of these conversations make me realize that sometimes we all get too focused on the details of the method without backing up and determining how they fit into the big picture. In this month's column, let's look at a couple of examples of this.


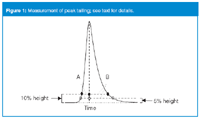
Figure 1
Fronting Peaks
In theory, at least, a chromatographic peak should be Gaussian in shape, with no fronting or tailing. Nearly every method, however, has peaks that exhibit some degree of peak tailing, where the back edge of the peak does not reach the baseline as quickly as the front edge rises from it. We measure peak tailing as either the asymmetry factor As or the tailing factor, TF. These are calculated as indicated below with reference to Figure 1:
As = B/A
TF = (A+B)/2A
where A and B are the peak half-widths, measured at 10% of the peak height for the asymmetry factor and 5% of the peak height for the tailing factor. In past years, methods were plagued with tailing peaks, especially when basic compounds were analysed on the older, low-purity, type-A silica columns. These columns had a high population of acidic silanol groups responsible for tailing. Today's newer, high-purity, type-B silica columns are much less prone to tailing. In fact, sometimes tailing is so small that we begin to notice peak fronting.
One conversation related to peak fronting of 0.8, as measured with the asymmetry factor. The person was very concerned about the source of the fronting and how to correct it. It is easy to get off on a conversation about the sources of fronting peaks, which, in general, are rare today with reversed-phase methods. Peak fronting is most commonly attributed to a gross column failure that we sometimes refer to as bed collapse. Some deterioration of the packing material takes place and the particles inside the column shift, creating a void in the column. This will cause all the peaks in the chromatogram to front, and will not be corrected by column reversal or flushing. The lady had replaced the column with a new column twice and the fronting persisted, so it is unlikely that column collapse was the problem. Another possible problem source is insufficient buffer in the mobile phase. Also, in the past I have seen references to peak fronting in ion-pairing separations being corrected with a change in temperature, but these methods used type-A columns; I have not seen this on type-B columns, so a temperature change may no longer be effective.
As the conversation became more involved, one of my colleagues, who was listening, waved the yellow caution flag. "Wait a minute," he said. "How much fronting are you seeing?" Well, a little simple math says that an asymmetry factor of 0.8 is equivalent to the same peak distortion as a peak tail of 1.25. The release specifications that many column manufacturers use in their quality testing process indicate that a column with 0.9 < As < 1.2 is acceptable. In other words, a brand new column might exhibit a little fronting or tailing. If the method we were discussing had an asymmetry factor of 1.25, we wouldn't be having this conversation, would we? As my daughter used to say, "Don't sweat the petty stuff (...and don't pet the sweaty stuff)." This is not a problem that is worth investigating.
Excessive Recovery
Another Pittcon attendee dropped by to discuss a problem he was having with a method for the analysis of a drug in serum. When he calculated recovery of the drug from spiked samples, he found 102% recovery. Having low recovery, for example, 98%, is easy to explain, but he was concerned about having too much recovery — how is it possible to recover more drug than you put in? We must remember that errors in most laboratory processes, including sample preparation and chromatography, are distributed evenly about the mean. For example, it is just as likely that a pipette will deliver 0.5% more than the nominal value as it is to make the same error on the low side. As a result, the overall error of the method should be distributed about the mean value. However, with most sample preparation processes, we lose sample along the way, so the average recovery is <100%. For example, if the average recovery were 96 ±2%, we would never see >100% recovery, so when we do see a method with >100% recovery, we might be surprised. But there is nothing abnormal about such values.
As the conversation went along, questions centred on the source of the error. It turns out that the method recovery was measured by comparing extracted serum samples with an aqueous reference standard. While this is a reasonable technique to make a gross check of overall extraction efficiency, it is not appropriate for method calibration. With bioanalytical methods (drugs in biological matrices), the regulatory guidelines call for a matrix-based standard curve. This means that the current method should use blank serum as the matrix and spike it at the appropriate concentrations to generate the calibration curve. This provides some internal correction for some of the variables that might be beyond the control of the user. For example, ion enhancement or ion suppression with mass spectrometric (MS) detectors can be a problem with serum- or plasma-based methods. By using a matrix-based standard curve, in which the calibrators are treated the same as the samples, it is much like solving simultaneous equations in algebra — the constant factors drop out.
But up comes the yellow flag again! Why are we having this conversation? The regulatory guidelines for methods like this allow for precision and accuracy of ±15% at all concentrations above the lower limit of quantification (LLOQ) and ±20% at the LLOQ. A 2% error, as in the present case, is insignificant relative to the allowable variability. There are other fish to fry.
The Lake Wobegon Effect
I am reminded of a story told to me by a colleague in a laboratory I used to manage. He had worked for a major pharmaceutical company that, like most pharma companies, was very interested in improving their processes. As part of the data-gathering process, when each new LC method was completed, the time taken to develop the method was added to a database. After a sufficient amount of data was gathered, it was possible to calculate the average method development time for an LC method. All was well and good until the next method was developed and it took longer than the average to complete. The staff was chastised for poor performance because the laboratory manager expected all methods to be developed in less than the average time. This reminds me of Garrison Keillor's mythical town of Lake Wobegon that he describes on his National Public Radio broadcasts. The tag line at the end of his Lake Wobegon news always ends with "...and all of the children are above average." If the laboratory had sufficient data to determine a statistically significant average, it is unreasonable to expect all methods to be developed in less than the average time — duh! Now if the average represented all LC methods developed in the pharmaceutical industry, one company's goal of developing methods in less than the average time might be reasonable. Or in a continuous-improvement environment, it might be reasonable to expect a target development time to be less than one standard deviation above the mean. But all methods less than the average time? What are they smoking?
Count the Cost
Another place where we can get distracted from the big picture is related to trying to reduce analysis costs. My guess is that I get this question in at least half of the LC classes that I teach: "How can I extend the life of my column?" Whenever we are looking at trying to improve a method, we need to consider the cost. How much does the current status cost? How much can I save with the desired change? How much will it cost me to get there?
Unfortunately, many laboratories consider the purchase of an LC column to be a capital expenditure. Yes, it is expensive, but in terms of the overall cost of analysis, it should be considered a consumable item. Do a few calculations and you'll convince yourself (and hopefully your boss). When I was managing a contract analytical laboratory, we were often asked for a quote for budget purposes. For a typical LC method with ultraviolet (UV) detection, we used a number of $50/sample for this purpose. If I pay $500 for a column and only get 500 samples through it before it fails, the cost is $1/sample for the column. This is 2% of the overall cost of the method in the present example. A 500-injection lifetime is pretty short for most methods, so you might be prompted to spend some time trying to increase the column lifetime. Let's say that you do some experimentation and find that by instigating a special cleaning procedure, you can extend the column lifetime to 1000 injections. Well, you've just cut your column costs in half. This sounds pretty good until you consider the overall savings. You've reduced the column burden on the method from 2% to 1%. Is it worth the trouble? Instead, it might have been more appropriate to focus on a more expensive part of the process — maybe it is report generation or sample tracking. There are enough things to take up our time in the laboratory without creating new ways to spend time that have little return on the overall value of the process.
The Big Picture
So, what's the common thread with these stories? It is very easy to get focused on one specific aspect of an LC method and get distracted from the overall goal of the method.
In the first case, why is peak asymmetry of 0.8 a concern? If it is because we're not used to seeing fronting peaks in most LC methods, we might be worrying about nothing. If it is because we're concerned about losing resolution between that peak and a small peak that is eluted just in front of it, then the concern might be more valid. Should we focus on peak fronting or on adjusting the relative peak positions?
In the second example, recovery of 102% turned out to be unimportant in the context of a bioanalytical method with allowable precision and accuracy of ±15–20%. But if the method were a pharmaceutical content uniformity assay, in which ±2% is the allowable variation, 102% recovery is a real concern.
Continuous improvement, including the reduction of method development time, is an admirable goal. But is it reasonable to instantly expect all methods to be better than average? Setting more achievable intermediate goals for method improvement would be more reasonable, more likely to succeed, and certainly better accepted by the method development staff.
These examples bring to mind one of my favorite authors when I was managing a laboratory: Eliyahu Goldratt (The Goal; It's Not Luck). In what some people refer to as a BFO (blinding flash of the obvious), Goldratt introduced me to the concept of the bottleneck. You can spend all the time you want trying to improve a process, but if it does not affect the rate-limiting step, your efforts will be of little help. Instead, if you can improve just this one step, the whole process will be improved. This principle applies very easily to the LC laboratory. Don't spend too much time investigating insignificant aspects of a method — focus on what will really make a difference.
"LC Troubleshooting" editor John W. Dolan is vice president of LC Resources, Walnut Creek, Califorina, USA; and a member of LCGC Europe's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting", LCGC Europe, Poplar House, Park West, Sealand Road, Chester CH1 4RN, UK, or e-mail Alasdair Matheson, the editor, at amatheson@advanstar.com


: Introduction and Liquid Chromatography")





; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Determining Enhanced Sensitivity to Odors due to Anxiety-Associated Chemosignals with GC
May 8th 2025Based on their hypothesis that smelling anxiety chemosignals can, like visual anxiety induction, lead to an increase in odor sensitivity, a joint study between the University of Erlangen-Nuremberg (Erlangen, Germany) and the Fraunhofer Institute for Process Engineering and Packaging (Freising, Germany) combined behavioral experiments, odor profile analysis by a trained panel, and instrumental analysis of odorants (gas chromatography-olfactometry) and volatiles (gas chromatography-mass spectrometry).


Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.
. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


