Too Many Changes
Remember the rule of one - change just one thing at a time.
Remember the rule of one — change just one thing at a time.
Each week I get emails from various readers with questions or problems (see contact information at the end of this column). I enjoy most of these and often they give me fodder for one of these "LC Troubleshooting" columns. This month I'd like to look at one of those problems, because it can give us some insight into the effects that certain changes will make with a liquid chromatography (LC) method.
The question went something like this: "I have a method that works well, but I am trying to scale it down so that I can save acetonitrile. The method uses a 150 mm × 4.6 mm, 5 μm particle C18 column operated at 2 mL/min at 30 °C. This is an isocratic method run with 40% acetonitrile and 60% of 0.1% formic acid. My sample is dissolved in mobile phase and I inject 20 μL. My peaks come out at 1.5 and 1.65 min, with an overall cycle time of 4 min. The resolution requirement of Rs > 2.0 is obtained easily. I decided to switch to a smaller diameter column to reduce the acetonitrile consumption. So I switched to a 150 mm × 2.1 mm, 5 μm C18 column that I found on the shelf and dropped the flow-rate to 1 mL/min, because the column is about half the diameter of the original one. Now I can barely pass the resolution requirements and sometimes fail. The pressure is higher than before and the peaks come out earlier. This seemed so simple, but I must have done something wrong. Can you help me?"
The Rule of One
This problem is a classic example of violation of the rule of one, which states, "change just one thing at a time." This is the scientific method, and we should use it to help identify the cause–effect relationship of changes we make to the system. As I see it, the column size, flow-rate and injection volume have changed and perhaps the column chemistry has, as well. Let's look at some of the factors that we should consider when making a change such as the one mentioned previously.
First, we need to make sure we have not made a chemistry change in the system. Based upon the question, I'm not sure if the column chemistry is the same between the two columns. There was a time when everyone thought that all C18 columns were created equal, but today, with literally hundreds of C18 columns to choose from, it might be more surprising if two are chemically the same than if they are different. The 2.1 mm column should be from the same brand and line of packing material as the original 4.6 mm column. Because this wasn't mentioned specifically in the question, I want to make sure it is not overlooked. A second way the chemistry of the column can change is if it has been used for other samples. A column that was "found on the shelf" might or might not be new. If it is used, it still might be OK to use, but this decision should be based upon a column log sheet that records column history and column testing. Any column with unknown history should, in my opinion, be filed in the dumpster. Columns are consumable items with finite lifetimes, and it isn't worth the risk of creating problems with a method by using a column with unknown history. Either of these changes, a different manufacturer's C18 material or a used column, can mean a change in the column chemistry and, thus, a possible change in peak spacing — one of the possible reasons the resolution requirements are hard to meet with the new column. For the moment, let's assume that the 2.1 mm column was from the manufacturer and packing type and was new or like-new.
Scaling the Column
The process of reducing the column diameter to save solvent is fairly simple, although there are some potential problems that should be kept in mind. As a guide, if the flow-rate is adjusted for the same linear velocity of mobile phase through the column, the retention times and column pressure should be the same with a column of different internal diameter. The flow-rate should be adjusted in proportion to the change in column cross-sectional area, which is proportional to the square of the internal diameter. So, in the present case, (4.6 mm/2.1 mm)2 = 4.8 ≈ 5. I usually use 5 as the factor, because it is easy to remember and I can do the calculations in my head. Thus, the flow-rate should be reduced from 2.0 mL/min to 0.4 mL/min for the smaller column. The retention times should be about the same as the original method, as should the pressure. Note that the proposed change was from 2.0 to 1.0 mL/min. This would give a relatively larger flow rate by a factor of a little more than two-fold and would result in shorter retention times and higher pressures, as observed. As a first step, I would lower the flow-rate to 0.4 mL/min to see if the results were comparable to the original conditions. Note, however, that the combination of changing the column diameter and flow-rate should not change resolution (ignoring extracolumn effects, see the following text), so this is not the source of the observed marginal resolution.
An increase in the relative flow-rate, especially with isocratic runs, can often be made with no penalty other than a higher pressure. Most of the time, conventional LC systems are run in the 2000–3000 psi (≈150–200 bar) range and are designed to perform well up to 6000 psi (400 bar), so higher pressure can usually be tolerated without ill effects. The critical measurement, in terms of selectivity, is the selectivity factor, α:


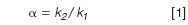
where k1 and k2 are the retention factors for two adjacent peaks, 1 and 2. The retention factor is calculated as follows:

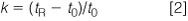
where tR is the retention time of a peak and t0 (sometimes called tM) is the column dead time. We can measure the column dead time from the unretained peak (often referred to as the solvent front or garbage peak), or we can estimate the column volume, VM as
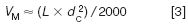
where L is the column length and dc is the column internal diameter, both in millimeters. VM is converted to t0 by dividing by the flow-rate.
Let's see where this leads us with the current method. First, we need to know the volume of each column. For the 4.6 mm column: (150 × 4.62 )/2000 ≈1.6 mL; for the 2.1 mm column: (150 × 2.12 )/2000 ≈ 0.33 mL. These convert to t0 values of (1.6 mL/2 mL/min) = 0.8 min, and (0.33 mL/1.0 mL/min) = 0.33 min, respectively. If the flow had been scaled properly, both columns would have t0 = 0.8 min. For the first peak in the original separation, k1 = (1.5 – 0.8)/0.8 = 0.875; the second peak, k2 = (1.65 – 0.8)/0.8 = 1.0625. This converts to α = 1.0625/0.875 = 1.21. Note that when the flow-rate is changed, tR for all peaks and t0 changed proportionally, so k and, thus, α stay constant. In other words, changing the flow-rate in isocratic separation, either directly or indirectly by changing the column diameter, makes no change in the peak spacing, or selectivity. This means that we can increase the flow-rate and reduce the run time, with the major observation being an increase in system pressure. There can be a minor reduction in column efficiency with real samples, but most isocratic methods can stand a two-fold change in flow without compromising resolution. What an inexpensive way to increase throughput! (Gradient separations require some compensating changes when flow is changed or selectivity will change).
Scaling the Injection
One thing that is often overlooked, and certainly was in the present case, is that the injection might need to be scaled with the column volume. If too large an injection volume is used, band spreading can occur on the column; if too large a sample mass is injected, sample overload can occur. We don't want either of these situations, so it is best to scale the injection with the change in column size. As a general rule, we can inject ≈15% of the peak volume of the first peak of interest without problems if we use the injection solvent as the mobile phase. The peak volume can be determined by drawing tangents to the sides of the first peak of interest and measuring the width between the two tangents where they meet the baseline. I didn't get a chromatogram for the present example so we can estimate the peak width. With reasonably well-behaved "real" samples, a 150 mm-long, 5 μm particle column should generate ≈10000 plates. The plate number, N, is calculated as
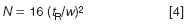
where w is the peak width at baseline. If we rearrange Equation 4 and solve for w, we get
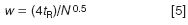
For the original method and the first peak, w = (4 × 1.5)/100 = 0.06 min. Convert this to volume by multiplying by the flow rate (0.06 min × 2 mL/min) = 120 μL. If we use our 15% rule of thumb for the injection volume, 15% × 120 μL = 18 μL. This tells us that the 20 μL injection in the original method is at the upper limit of the suggested injection volume. This might or might not be OK — it would be wise to see if the separation deteriorates if the injection volume is increased by two-fold. It is good to have a tested safety margin.
When the column is scaled down, the peak volume will change, too. If the flow is scaled properly, we would use 0.4 mL/min with the 2.1 mm i.d. column. The peak width in time should be the same, but the volume will be (0.06 min × 0.4 mL/min) = 24 μL, which is 1/5 of the original — in exact proportion to the change in the column volume. However, a 20 μL injection is almost as large as the peak width and will probably result in a broad peak, because the first molecules entering the column will have travelled a significant way down the column before the last molecules arrive at the top of the column. This increased peak width might be the reason that the resolution requirements of the proposed method were difficult to attain. A properly scaled injection would result in 15% × 24 μL = 3.6 μL. I would probably start at 5 μL and see if I could get away with it, then inject 10 μL and 2 μL to see if 5 μL had some safety margin before changes in resolution were observed.
When considering injection effects, it is the early peaks in the chromatogram that will be affected most strongly. As a general rule, we like to see the peaks fit in a retention window of 2 < k < 10, but if the separation has very many peaks, this might be hard, so we open the window to 1 < k < 20. When the retention factor is much less than 1, several potential problems can occur. Injection effects are more prominent for early peaks, the peaks are more likely to have interference problems with the tail of the unretained junk peak present in most samples and often resolution won't be as robust. In the present example, the peaks are in this danger region, which might be the reason why selectivity has changed when the column size was changed.
Extracolumn Effects
A final problem area to consider with the present method is extracolumn effects. This refers to any band broadening that takes place outside the column. The normal contributors to this are the connecting tubing between the autosampler and column, and between the column and detector, the injection volume and solvent, the detector cell volume and the detector time constant and data system data rate. The detector cell volume might not be changed easily, but when 2.1 mm i.d. columns are used, it is best to use 0.005 in. (0.125 mm) i.d. tubing in short lengths, keep the detector time constant no more than 1/10 of the peak width and make sure that the data rate is fast enough to collect at least 20 points across each peak. Keep the injection volumes small, as discussed previously, and try not to use an injection solvent that is any stronger than the mobile phase. Any peak broadening resulting from extracolumn effects will reduce resolution.
Summary
Reduction of the column diameter can be a very effective way to reduce solvent consumption. In the present example, when properly scaled, the amount of acetonitrile used would be reduced to 20% of the original method. An added benefit is that peaks will be narrower and, thus, taller (for the same mass on column), which generally improves the method performance near the detection limits. If you scale the flow-rate with the change in cross-sectional area, the retention times and pressure should stay constant. (It might be possible to reduce the run time by increasing the flow-rate with isocratic methods, but this is a factor that is independent of the column diameter.) When changing to a smaller diameter column, be sure to check for, and adjust if necessary, the proper injection volume. Also, be careful about other extracolumn effects, which become magnified as the peak volume is decreased with smaller-volume columns.
Erratum
In the July/August issue, LCGC Europe, 22(7), 360, Table 1, in the third column, the next to last line should read 403913, not 36827, and the 36837 should shift down one row. We apologize for the mistake.
"LC Troubleshooting" editor John W. Dolan is vice president of LC Resources, Walnut Creek, California, USA; and a member of the Editorial Advisory Board of LCGC Europe. Direct correspondence about this column to "LC Troubleshooting", LCGC Europe, Park West, Sealand Road, Chester CH1 4RN, UK.
For an on-going discussion of LC Troublshooting with John Dolan and other chromatographers, visit the Chromatography Forum discussion group at www.chromforum.org








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Determining Enhanced Sensitivity to Odors due to Anxiety-Associated Chemosignals with GC
May 8th 2025Based on their hypothesis that smelling anxiety chemosignals can, like visual anxiety induction, lead to an increase in odor sensitivity, a joint study between the University of Erlangen-Nuremberg (Erlangen, Germany) and the Fraunhofer Institute for Process Engineering and Packaging (Freising, Germany) combined behavioral experiments, odor profile analysis by a trained panel, and instrumental analysis of odorants (gas chromatography-olfactometry) and volatiles (gas chromatography-mass spectrometry).


Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.
. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


