Therapeutic Diazepam Monitoring in Human Plasma and Urine by HPLC: An Application for Alcoholism
LCGC Europe
A rapid and simple high performance liquid chromatography (HPLC) method with basic extraction assays was developed to investigate free diazepam levels in the plasma and urine samples of patients medicated with this drug for the management of alcohol withdrawal syndrome. The HPLC analysis was optimized and evaluated for linearity, imprecision, recovery, detection and quantification limits. The method showed linearity between 50–500 ng/mL (r2 ≥ 0.990). Coefficients of variations (%CV) were calculated to be in the range of 1.77–9.60. According to ICH guidelines, theoretical limits of detection (LOD) and quantification (LOQ) for plasma and urine were calculated as 8.3 ng/mL, 27.5 ng/mL and 8.2 ng/mL, 26 ng/mL respectively. Diazepam monitoring in plasma and urine displayed remarkable variations. The importance of adjusting doses according to individual requirements and the routine monitoring of plasma or urine for patients under medication is highlighted.
The 1,4-benzodiazepines constitute an important class of psychotherapeutic agents known for their hypnotic, tranquilizing and anti-convulsant properties. Comprehensive research has been performed to study their clinical performance.1 Diazepam is one of the most frequently prescribed benzodiazepines for the treatment of anxiety, sleep disturbance and alcohol withdrawal.2 However, the use of diazepam for alcohol withdrawal deserves clinical attention and monitoring because hepatic dysfunction usually accompanies alcohol dependence syndrome that depends on the metabolism rate in the liver.3
For the clinical, toxicological and biopharmaceutical study of benzodiazepines various biological fluids have been analysed. Clinically effective doses of benzodiazepines undergo extensive metabolic reactions and many of these metabolites are pharmacologically active.4
Identification of those pharmacologically active benzodiazepines is best performed on blood following a positive urine screening. It is, therefore, essential that assay methods are sensitive and specific (i.e., capable of separating and determining the parent drug, as well as its major metabolites).5
Various methods exist in the literature for the analysis of benzodiazepines in biological matrices.6–8 Immunoassay procedures [such as enzyme multiplied immunoassay technique (EMIT)/cloned enzyme donor immunoassay (CEDIA)] are widely applied methods for investigating addiction and poisoning.9
However, these immunoassay methods are not capable of differentiating the primal molecule from minor metabolites, so they are often applied together with other chromatographic techniques.
The use of gas chromatography–mass spectrometry (GC–MS) for benzodiazepine analysis has been reviewed11 and further studies have also been published describing other chromatographic applications.8,12 High performance liquid chromatography (HPLC) has also been studied5,13 and reviewed.14 Ease of extraction and application make liquid chromatography (LC) a reliable method,15–17 and attention has shifted to the development of HPLC methods to determine benzodiazepines in body fluids.11



In Brief
The extraction procedures are relatively simple, derivatization is not necessary and operation at ambient temperature allows the determination of thermally stable benzodiazepines. Strong absorption in the 230–260 nm wavelength region gives sensitivity in the nanogramme range and linearity over a wide concentration range. Moreover, because the technique is non-destructive, the eluted drugs can be recovered for further examination.18 In recent years, the coupling of LC to MS (LC–MS) has provided a useful and rugged technique for the analysis of drug compounds and an alternative to GC–MS in which some compounds thermally decompose to give metabolites common to many of benzodiazepines.19–21 Now, LC–MS is a fairly established technique for benzodiazepine analysis.22,23 Compared with LC–UV systems, LC–MS is much more selective and sensitive. In applications where plasma concentrations are high, the use of the LC–UV is more than adequate. Whereas, for clinical purposes LC–MS is quite expensive where low costs are mandatory for routine clinical analysis. Because HPLC-UV assays are inexpensive and widely used, it appeared to be the best option to perform simultaneous separation, quantification and clinical monitoring of diazepam as a primary concern of this paper.
As well as clinical research applications, some other applications involving the determination and quantification of benzodiazepines with HPLC-UV analysis technique have also been reported recently.24,25
Differences in the metabolism rates of benzodiazepines in humans can be expressed by monitoring drug levels.4 When considering the management of alcohol withdrawal, as well as the clinical assessments, the dose of diazepam and free diazepam levels in biological fluid is required to display the variations in half-life of benzodiazepines according to each patient.26
The scope of this study is to construct and validate a rapid and simple method to determine and monitor free diazepam in human plasma and urine; and also to emphasize the correct dosage of diazepam by simultaneously monitoring its levels in plasma and urine. For this purpose two groups of patients being treated for alcohol-withdrawal syndrome in Ankara University Hospital Psychiatry Clinic were gathered. Free diazepam levels in the plasma and urine samples from patients in the first group who had single oral dose of diazepam, were investigated to display the personal variations of drug metabolism. From the same point of view, the findings from the second group who had repeated daily constant doses were investigated, to denote drug accumulation in individuals because of metabolism variations.
Experimental
Chemicals and reagents: Sodium carbonate, sodium dihydrogen phosphate dehydrate, disodium hydrogen phosphate, sodium hydroxide, ethyl acetate, diethyl ether and HPLC-grade methanol were purchased from Merck (Darmstadt, Germany). The standards of pharmaceuticals, diazepam and nitrazepam (Internal standard) were obtained from Sigma-Aldrich (St Louis, Missouri, USA). Prescribed diazepam tablets for patients were obtained from pharmaceutical firm Deva Medical Company (Istanbul, Turkey). All chemicals were of analytical-grade in the highest purity available.
Instrumentation and Chromatographic Conditions
The quantification of diazepam was performed by high-performance liquid chromatography (HPLC), equipped with a UV detector. HPLC analysis was performed using an HP Agilent 1100 (Santa Clara, California, USA). The system consisted of an isocratic pump, a manual injector with a loop volume of 20 μL, and a Zorbax SB C18 column with diameters 5 μm, 4.6 × 250 mm. The wavelength at 230 nm was chosen for UV detection.
The mobile phase (methanol, phosphate buffer and water, 70:20:10, adjusted to pH 7.5) was filtered through a 0.45 μm membrane (Alltech, Illinois, USA) and degassed in an ultrasound bath for 30 min. An isocratic solution was performed at a flow-rate of 1.0 mL/min at room temperature. Peak areas were measured and calculations were performed considering the internal standard (IS) peak ratios.
Preparation of Stock Solutions and Working Standards
Nitrazepam was chosen as the IS due to its similar physico-chemical properties with diazepam. Stock solutions of diazepam and nitrazepam (IS) were prepared in methanol solution at the concentrations of 10 μg/mL and 2 μg/mL respectively. Working standards of diazepam, used for spiked plasma and urine samples, were prepared weekly in the concentrations of 0.5, 1, 2, 3, 4 and 5 μg/mL and made by the dilution of the stock solutions with methanol.
Blank human blood samples were collected from Ankara Turkish Red Crescent Blood Centre and centrifuged at 2000 g for 5 min to separate the plasma. Blank urine samples were collected from the laboratory crew of Institute of Forensic Medicine, Ankara University. Plasma and urine samples, and all working solutions, were stored at –20 °C until the analysis was performed, and were checked chromatographically for purity before experiments, were used as quality control (QC) specimens and were checked for the stability before and after the injections of every sample set.
Sample Preparation
Blank plasma and urine samples (800 μL) were placed into clean glass tubes containing 2 μg/mL IS (100 μL) solution. Each of these tubes 100 μL of diazepam solutions were spiked in the concentrations of 0.5, 1, 2, 3, 4 and 5 μg/mL reaching the total volume to 1 mL and achieving 10 times diluted mixtures; yielding analyte concentrations as calibration samples namely 50, 100, 200, 300, 400, 500 ng/mL diazepam and 200 ng/mL nitrazepam. Different from the calibration solutions, 100 μL methanol was added instead of diazepam solution for the samples from the patients.
Extraction: Liquid–liquid extraction (LLE) was applied for plasma samples.5 A volume of 2 mL of diethyl ether was added to the samples, vortexed for 1 min. and centrifuged at 3000 g for 5 min. Extraction procedure was repeated twice. The organic phase (diethyl ether) was collected to another sample tube and dried under nitrogen. Remaining analyte was dissolved with methanol to be ready for injection. For urine samples pH is adjusted to 9.5 with addition of sodium carbonate before the same extraction procedure was applied. As organic solvent, ethyl acetate was preferred instead of diethyl ether.
Patients
Sixteen male patients from Ankara University, Psychiatry Clinic, Alcoholism Section participated in this study. The average age of participants was 49 ± 9. All had at least a 15-year addiction to alcohol and had normal values from hepatic function tests. Participants were separated into two groups. The first group, consisting of 12 patients, was medicated with a single oral dose of 10 mg diazepam as an introductory treatment of alcoholism. This second group, consisting of four patients, was medicated for three days with a repeated daily oral dose of 10 mg diazepam. After each administration, blood and urine samples were collected at 2 h and 4 h respectively. In this article, to clearly demonstrate the distinction between groups, the first group of patients were represented with numbers where as the patients in the second group were represented with letters.
This study was approved by the Ethical Committee of Ankara University, Faculty of Medicine.
Results and Discussion
The HPLC method appeared rapid and simple.
Validation of Methods
The procedures and HPLC method developed to determine diazepam from plasma and urine were fully validated according to the following criteria.
Specificity and selectivity: Methods for plasma and urine both demonstrated excellent chromatographic specificity with no endogenous interference at the retention times of the IS and diazepam (5.8 and 8.7 min. respectively). A representative chromatogram for human plasma spiked with Diazepam (500 ng/mL) and the IS (200 ng/mL) is shown in Figure 1.

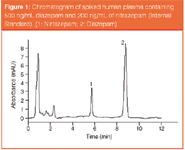
Figure 1: Chromatogram of spiked human plasma containing 500 ng/mL diazepam and 200 ng/mL of nitrazepam (Internal Standard). (1: Nirtazepam; 2: Diazepam)
Chromatograms of both urine and plasma samples for the monitoring of diazepam levels of a patient are displayed in Figure 2. The UV detection was set to optimum wavelength of 230 nm; above this wavelength nitrazepam signals show a continual increase in sensitivity; however, diazepam signals begin to decrease in sensitivity.

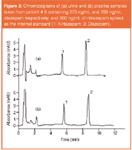
Figure 2: Chromatograms of (a) urine and (b) plasma samples taken from patient # 8 containing 273 ng/mL and 289 ng/mL diazepam respectively; and 200 ng/mL of nitrazepam spiked as the internal standard (1: Nirtazepam; 2: Diazepam).
Linearity: After establishing the chromatographic conditions, separate calibration curves were prepared for plasma and urine over a diazepam concentration range of 50–500 ng/mL. For each concentration five individual replicates were injected and linearity was obtained for both methods with the correlation coefficients (r2) over 0.990.
Precision: Precision, defined as relative standard deviation (RSD) or coefficient of variation (CV), was determined by five individual replicates at three different concentrations (n = 5). Table 1 shows the CV values of concentrations 50, 300 and 500 ng/mL as low, medium and high concentrations respectively to present intra-day precision.


Table 1: Confidence parameters of validated methods for the determination of diazepam in plasma and urine samples by (n = 5).
Recovery: The recovery of extraction procedures from human plasma and urine were determined by comparing pre-extraction spikes with the post-extraction spikes of the IS. Three individual replicates of spiked samples at mid-concentration (300 ng/mL diazepam) were prepared with and without an internal standard (n = 3). The extraction procedure was conducted as described previously. Peak area ratios were compared and recoveries were calculated as 89% and 77% for plasma and urine respectively.

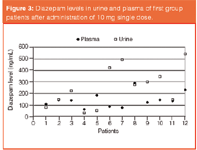
Figure 3: Diazepam levels in urine and plasma of first group patients after administration of 10 mg single dose.
Limit of detection and quantification: Limit of detection (LOD) and lowest limit of quantification (LOQ) were determined based on the standard deviation of the response and the slope of the calibration curve, according to ICH guidelines27 (LOD = 3.3σ/S, LOQ = 10σ/S where σ is the standard deviation (SD) of the response and S is the slope of the calibration curve). LOD and LOQ values were calculated both for plasma and urine samples and found as 8.3 ng/mL, 27.5 ng/mL and 8.2 ng/mL, 26 ng/mL respectively.

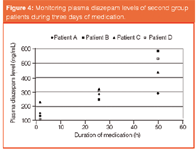
Figure 4: Monitoring plasma diazepam levels of second group patients during three days of medication.
Diazepam Levels of Patients
Considerable differences for each individual were observed. Variations in the diazepam levels detected in the first group of patients are shown in Figure 3. Even though each patient was medicated with an equivalent single oral dose, diazepam levels in plasma and urine were quite dissimilar. Variations may arise because of the enzyme differences among individuals. In this respect, for the application of an effective dosage, the enzyme polymorphism should be taken under consideration in medical therapies. Furthermore, in the second group of patients, accumulation of diazepam was expressly monitored. Figures 4 and 5 show that the diazepam levels in plasma and in urine both increase as the medication continues with repeated equal doses. It may also be explained by the differences of metabolism rates of individuals. Diazepam is metabolized mainly by cytochrome P450 isoform CYP2C19. The resulting data may also be considered as a pre-research for pharmacogenetic polymorphism in this group of alcoholics.

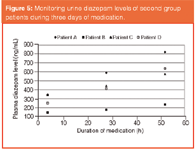
Figure 5: Monitoring urine diazepam levels of second group patients during three days of medication.
Conclusions
As well as monitoring the diazepam concentration in plasma and urine, this study also aims to analyse the drug's benefits for alcoholics from a psychological perspective. As the free diazepam level increases in biological material, the expected medical effects also increase. The optimum drug use with maximum results is the main objective for psychiatrists. It is recommended that doses of benzodiazepines should be determined depending on the severity of the symptoms.28 The monitoring of benzodiazepine levels during alcohol detoxification would help to control the withdrawal symptoms alongside other clinical methods used to assess the severity of psychological alcohol withdrawal symptoms.29,30
The primary concern of this study is simplicity for the method to be used in clinics or hospitals. Therefore, rather than the method itself, this study stands out for its clinical application. And combining the different perspectives of clinicians and analytical chemists forms the main feature of this research. Although traditional clinical applications do not use analytical instruments for treatments such as alcohol withdrawal, this study showed that the dosage of medication should be adjusted carefully according to the analytical data relating to drug levels in plasma and urine for each individual patient.
Acknowledgement
This study is supported by grants from Ankara University Research Fund with the project of 20.010.000.004 "Toxicological Analysis of Abused Drugs"
Görkem Mergen obtained his PhD in Forensic Chemistry and Toxicology at the Institute of Forensic Medicine, Ankara University in 2008. He has been working as RA and TA at the same institution since 2002. His research interests are toxicology, chromatography and spectroscopic methods in terms of toxicological analysis. He mainly focuses on analytical method development that can be applied to biological tissues, which also includes post-mortem materials.
Tülin Söylemezoglu is a professor and the chairperson at the same institution since 1994. She gained her PhD degree in toxicology from the Faculty of Pharmacy, Ankara University at 1974. She lectured in the Faculty of Pharmacy, Medical School and Institute of Forensic Medicine. Her expertise and research areas are analytical toxicology, drug abuse, free radical toxicology and genetic toxicology.
Inci Ilhan is an associate professor in the Psychiatry Clinic, Alcoholism Section of the faculty of Medicine, Ankara University. She has been working with alcoholics in the same department as a psychiatrist and she has been performing research on alcoholism and cures for addiction.
Yildirim B. Dogan is a professor of psychiatry in the same institution. He had built his academic career in medicine at Ankara University and University of London. He is an expert in adult psychiatry and addiction psychiatry. He heads the alcoholism section concerned with research areas both in alcoholism and addiction.
References
1. T.R. Norman and G.D. Burrows, Neuro-Psychopharmacol. & Biol. Psyciat., 8, 115–126 (1984).
2. A.D. Ciraulo and P.E. Nace, Am. J. Addict., 9, 276–284 (2000).
3. M.P. Peppers, Pharmacotherapy, 16, 49–58 (1989).
4. A. Smith-Kielland et al., Scand. J. lab. Invest., 61, 237–246 (2001).
5. T.B. Vree et al., J. Chromatogr. B, 162, 605–614 (1979).
6. A. Sioufi and J.P. Dubois, J. Chromatogr. B, 531, 459–480 (1990).
7. O.H. Drummer, J. Chromatogr. B, 713, 201–225 (1998).
8. M. Rizzo, J. Chromatogr. B, 747, 203–216 (2000).
9. F. Divanon, D. Debruyne and M. Moulin, J. Anal. Toxicol., 22, 559–566 (1998).
10. A.W. Jones, A. Holmgren and P. Holmgren, J. Chromatogr. B, 580, 3–4l (1992).
11. H.H. Maurer, Forensic Sci. Int., 146, 1–7 (2004).
12. J.L. Valantine, R. Middleton and C. Sparks, J. Anal. Toxicol., 20, 416–424 (1996).
13. J.B. Lloyd and D.A. Parry, J. Anal. Toxicol., 13, 163–168 (1989).
14. C.A. Mehta, Talanta, 31, 1–8 (1984).
15. W.E. Lambert et al., J. Anal. Toxicol., 19, 35–40 (1995).
16. M. Rizzo et al., J. Chromatogr. B, 731, 207–215 (1999).
17. A.W. Abu-Qare, B. Mohammed and B. Abou-Donia, J. Chromatogr. B, 754, 503–509 (2001).
18. B. Kinberger and P. Wahrgren, Analytical Letters, 15, 549–557 (1982).
19. M. Tatsuno et al., Jpn. J. Toxicol. Environ. Health, 42, 248–256 (1996).
20. H. Essien et al., J. Chromatogr. B, 683, 199–208 (1996).
21. S. Mcclean et al., J. Chromatogr. A, 838, 273–291 (1999).
22. S.J. Marin et al., J. Anal. Toxicol., 32, 491–498 (2008).
23. E.I. Miller, F.M. Wylie and J.S. Oliver, J. Anal. Toxicol., 32, 457–469 (2008).
24. K.B. Borges et al., Talanta, 78, 233–241(2009).
25. Y. Li et al., Yiyao Daobao, 26, 1415–1417 (2007).
26. M. Lejoyeux, J. Solomon and J. Ades, Alcohol Alcoholism, 33, 563–575 (1998).
27. ICH. International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of Analytical Procedures: Methodology (ICB-Q2B) (1996).
28. J.T. Sullivan, R.M. Swift and D.C. Lewis, J. Clin. Psychopharm., 11, 291–295 (1991).
29. A. Foy, S. March and V. Drinkwater, Alcohol. Clin. Exp. Res., 12, 360–364 (1998).
30. A.M. Holbrook et al., Can. Med. Assoc. J., 160, 675–680 (1999).





















