The QuEChERSER Mega Method
Introduced in 2003, the “quick, easy, cheap, effective, rugged, and safe” (QuEChERS) sample preparation approach has been widely adopted in many applications, particularly in chemical residue analysis of foods. Prior to QuEChERS, sample preparation generally entailed several time-consuming, labour-intensive, and reagent-excessive steps, but the commercialization at the time of powerful, cost-effective, benchtop gas chromatography–mass spectrometry (GC–MS) and liquid chromatography–tandem MS (LC–MS/MS) instruments enabled the implementation of the QuEChERS procedure. Despite analytical technologies continuing to improve over the last two decades, many laboratories are still using QuEChERS protocols developed for outdated instrumentation. Recently, QuEChERS has been updated into QuEChERSER (with “efficient and robust” being added to the portmanteau) to better take advantage of the features provided by modern sample preparation and analytical techniques. Most notably, QuEChERSER is a “mega-method” that covers a broader scope of polar and nonpolar analytes in diverse sample types. In this article, the new QuEChERSER approach and its advantages over QuEChERS are discussed.
Twenty years ago, the “quick, easy, cheap, effective, rugged, and safe” (QuEChERS) approach for sample preparation was developed by Anastassiades and Lehotay at the United States Department of Agriculture–Agricultural Research Service (USDA‑ARS) Eastern Regional Research Laboratory (1,2). The goal was to develop the most efficient method for the multiclass, multiresidue analysis of pesticides in foods, which had been a long-standing wish for regulatory and industry laboratories for decades. However, increasing concerns, such as excessive glassware, use of chlorinated solvents, high labour needs, and low sample throughput, were putting pressure on laboratories to adopt more efficient practices. Simultaneously, more sensitive, smaller, and less expensive gas chromatography–mass spectrometry (GC–MS) and liquid chromatography–tandem MS (LC–MS/MS) instruments were also being introduced, which allowed for a broader scope of analysis while still maintaining high selectivity in detection. The introduction of these new GC–MS and LC–MS/MS instruments meant that sample preparation needed to recover a wide range of polar and nonpolar pesticides in the same method. Because chemical interferences were less likely to occur, cleanup could be “just enough” to reduce indirect matrix effects and avoid instrument contamination.
We decided to start from scratch, and QuEChERS essentially grew out of brute force experimentation of each aspect in the extraction and cleanup process (2). We eventually achieved the most streamlined protocol to yield high recoveries with sufficiently clean final extracts for moistened food samples. Acetonitrile was shown to work best for extraction. MgSO4/NaCl induced the most effectively tailored phase separation to avoid proteins and sugars, and dispersive solid-phase extraction (d-SPE) cleanup with a combination of anh. MgSO4, primary secondary amine (PSA), C18, and graphitized carbon black (GCB) sorbents worked well to retain lipids (including fatty acids) and chlorophyll. We realized that traditional SPE cartridges were expensive, inflexible, and inefficient in “chemical filtration” applications in which the extract also serves as the eluting solvent, so we decided to just combine the extract with the sorbents.
Two versions of QuEChERS were independently validated in successful interlaboratory trials for analysis of pesticide residues in foods (3,4). In 2010, Majors interviewed Lehotay and Anastassiades for LCGC to discuss the past, present, and future of QuEChERS (5). Since then, many reviews have been published about QuEChERS, including descriptions of how it has expanded beyond the monitoring of pesticides in foods (6–8). Lehotay and Chen (9) plotted the trend in scientific publications involving QuEChERS, tracking a total of 2565 papers on the topic through 2017. In a continuation of the same search in Web of Science, QuEChERS has grown from nearly 450 to >600 papers per year, totaling 4499 publications through 2020. More than 30 vendors worldwide market QuEChERS products, which has helped it become a staple approach in sample preparation. The chemistry of QuEChERS possesses a flexibility that enables it to be a template for modifications, while still maintaining streamlined protocols for a wide range of applications.
Despite the popularity of QuEChERS, analytical instrumentation has continued to improve in the past two decades, which enables even more streamlined sample preparation with broader applicability. For example, extract and inject methods are now feasible in many cases to overcome matrix effects using highly sensitive and selective MS-based analyses (10,11). However, robust cleanup is still often needed to provide long‑term ruggedness in high-throughput applications involving complex matrices.
Recently, an updated approach in streamlined sample preparation, dubbed QuEChERSER, has been introduced (12–16). QuEChERSER adds “efficient and robust” to the QuEChERS portmanteau, but more importantly, QuEChERSER is a “mega-method” that covers a wider polarity range than QuEChERS. The most effective way to gain laboratory efficiency is by reducing the number of methods needed to analyze the same scope of analytes. For example, four individual methods are typically used to monitor pesticides, environmental contaminants, veterinary drugs, and mycotoxins in pertinent foods, but with QuEChERSER, the same sample can be prepared by the same method to cover the contaminants in all kinds of foods.
This article describes and explains the changes made in QuEChERS leading to the development of the QuEChERSER method.
QuEChERSER vs. QuEChERS
Table 1 outlines each step in the QuEChERS and QuEChERSER templates for comparison, and the following discussion is sectioned by the individual components. Only the differences in bold text (Table 1) are discussed. Otherwise, both methods are streamlined to employ liquid extraction of sample test portions in tubes by shaking for 10 min followed by centrifugation for 3 min.


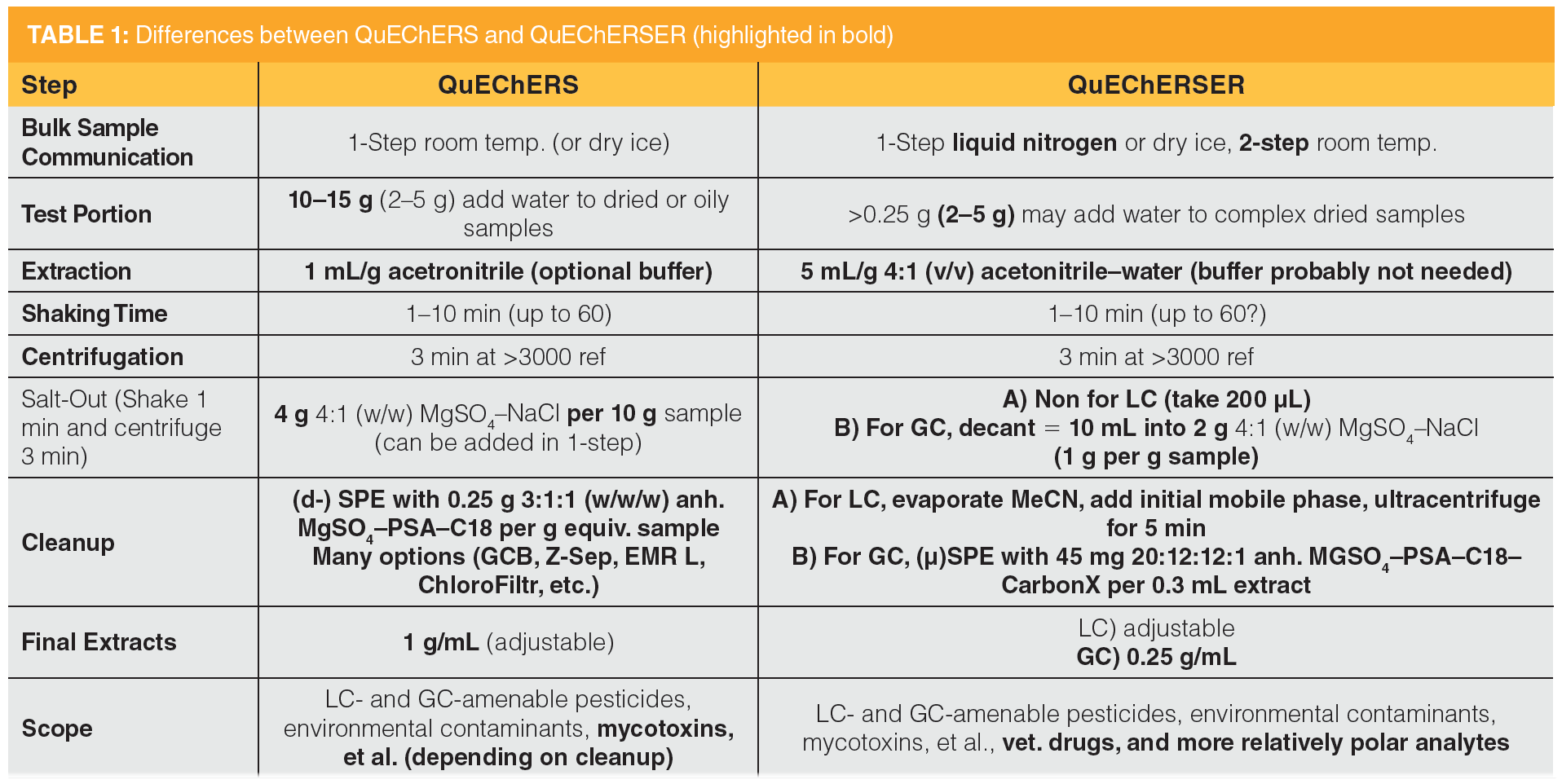
Sample Comminution: An easy way to increase practical efficiency in sample preparation is by reducing test portion sizes. However, sample processing (comminution) is rarely included by most chemists as part of the analytical method even though it is as equally (or more) important as any other step. As discussed in previous papers (9,12,17–20), comminution must not be ignored! Fundamentally, all analytical chemists must understand that ineffective sample comminution relative to the test portion size for analysis leads to a “garbage in, garbage out” situation. No matter how accurate the sample preparation and analytical methods are demonstrated to be based on the validation and quality control (QC) results, the expenses and effort put into the analyses are rendered meaningless if the analyzed test portion fails to represent the original sample.
Prior to QuEChERS, most methods called for 50–100 g test portions of bulk processed food samples in pesticide residue analysis, but studies showed that 10–15 g subsamples could yield similar results with better comminution practices, such as using two steps, high-quality devices, or cryogenics (18). In QuEChERSER, test portions can be further reduced to 1–5 g by the same concepts, but it is better to use liquid nitrogen rather than dry ice for reasons described in the literature (12,19). For QuEChERSER, a safe, facile, and high-throughput protocol was developed using conventional sample processors for (unfrozen) raw bulk food commodities applying liquid nitrogen in a single step. With practice, two technicians alternating two containers for a single device can comminute a batch of 40 bulk samples and weigh out the test portions in centrifuge tubes ready for extraction in approximately 4 h.
This type of efficient comminution using liquid nitrogen can be done for any sample preparation method to gain the benefits of reduced test portion sizes. Independent of the subsequent steps, the degree of subsample homogeneity can be empirically measured by adding a QC spike to the bulk samples prior to comminution. The repeatability and recovery of the QC spike is then determined for each batch of samples to isolate and assess the sample processing step, as well as similar QC spikes added just prior to the sample preparation and analysis steps to follow (12,20). The measurement uncertainty and contributions of each step in the overall method are then calculated using summation of squares to determine the limiting source of error for control and improvement in routine monitoring. Despite the smaller test portion, QuEChERSER has been demonstrated to yield about 5% lower overall relative standard deviation (RSD) (better reproducibility) than QuEChERS (12,20).
Extraction: Prior to QuEChERS, extraction typically called for 2 mL or more of the solvent. QuEChERS reduced this ratio to 1 mL/g sample for three reasons: 1) a greater volume could not fit well within the convenient 50 mL extraction tubes; 2) use of the smaller solvent-to-sample ratio led to more concentrated extracts for potentially lower detection limits (without solvent evaporation); and 3) a 1 mL/g ratio was determined to be acceptable for complete extraction of the tested incurred analytes in real samples (2–4). In the latter case, using more solvent per test would have been desirable to provide thorough extraction for all types of analytes and matrices, but the former two reasons precluded a change to the method, which is one of the reasons why QuEChERS is not ideal for extracting lipophilic analytes in relatively fatty matrices. Furthermore, analytical tools are capable of orders of magnitude lower detection limits nowadays than when QuEChERS was developed, thus more dilute extracts are no longer a problem, and in fact, dilution is often used to overcome matrix effects in MS analyses.
The reduction of the test portion size in QuEChERSER allowed for the reconsideration of the 1 mL/g ratio used in QuEChERS. Samples of 2 g in 15 mL centrifuge tubes can be readily extracted with approximately 10 mL of solvent (or 30 mL for 6 g test portions in 50 mL tubes). As it turned out, the multiclass, multiresidue method developed in my laboratory for veterinary drug residues employed 10 mL of a acetonitrile–water solution in a 4:1 (v/v) ratio for extraction of 2 g test portions of animal tissues (10,13,21). This 5 mL/g solvent-to-sample ratio has been shown to fully extract many incurred drugs in different matrices, and the USDA Food Safety and Inspection Service has been using this approach routinely in the National Residue Program for nearly a decade (22). In QuEChERSER, this proven extraction protocol was simply extended to also include pesticides, environmental contaminants, and other types of analytes in the same sample.
Another major factor for extraction in nearly all methods is the amount of water in the sample or extraction solvent. For dry commodities, such as grains, QuEChERS and other nonaqueous extraction methods require the addition of water to the test portions prior to extraction with the organic solvent. However, the extraction solvent already contains 20% water in QuEChERSER, thus the water addition step is avoided. QuEChERS for dry samples typically entails 2 g samples and 5 mL of water, followed by extraction with 10–15 mL of acetonitrile, which is not too different from the aqueous acetonitrile extraction conditions in QuEChERSER.
The limitation in QuEChERS with nonpolar analytes in fatty food matrices and soils was also addressed in QuEChERSER by an increased solvent‑to-sample ratio. Even though isotopically labelled surrogate standards are used in both methods to compensate for <100% recovery of legacy chlorinated pesticides and environmental contaminants in fatty foods, the accuracy of the QuEChERSER method is substantially better than with QuEChERS (13,16). Better cleanup using mini‑cartridge SPE rather than d-SPE prior to GC analysis is another major reason, and is discussed later in this article.
Precipitation Cleanup and Ultracentrifugation for LC Analysis: An advantage and disadvantage of QuEChERS is that the same final extract is usually used for both GC and LC analyses, which is a convenient and efficient benefit, but it also limits the performance and scope of the analyses, particularly for LC-only analytes. Hydrophilic analytes do not partition into the acetonitrile layer during the salting-out partitioning step, and in (d-)SPE, PSA and its alternatives retain many LC-amenable carboxylic acid analytes. Furthermore, the final extract in QuEChERS consists almost entirely of acetonitrile, which is not ideal for injection in reversed-phase LC. The first-to-elute analytes tend to have poor peak shapes even when the final extracts are partially diluted with water.
In QuEChERSER, a small aliquot (200 µL) of the initial extract before phase separation is taken for LC analysis, which expands the analytical scope to many polar compounds (such as many drugs) that do not partition into acetonitrile from water. This extract can be taken directly for LC analysis as done in extract and inject methods (10,13–14), but this runs into the same problem as in QuEChERS with poor peak shapes of the most polar analytes.
In the QuEChERSER method, a solution that also provides cleanup is to perform a solvent-exchange step that matches the final extracts with the initial mobile phase. A common evaporation device using nitrogen flow evaporates a batch of 200 µL extracts at 40 °C in approximately 5 min. Then, the initial mobile phase solvent is added (750 µL), which does not dissolve the hydrophobic matrix components that had precipitated from the extracts in the micro-centrifuge tubes. Nonpolar analytes also precipitate, but those are analyzed by GC anyway, which minimally affects the overall scope of analysis.
Otherwise, sorbent-based cleanup tends to lose certain analytes, which was shown in a recent comparison study involving four veterinary drug methods (23). Similarly, in the case of ultrafiltration, adsorption of some analytes always occur in mega-methods for any one type of membrane material, especially in a highly aqueous solvent as in this application (24). Furthermore, exposure of final extracts to filters and sorbents sometimes adds more potential interferants in the analysis than they remove. Thus, QuEChERSER employs a 5 min ultracentrifugation (13,000 relative centrifugal force) step after precipitation cleanup to maintain the exceptionally broad scope of analysis. Although vendors who sell sorbents and filters do not promote precipitation cleanup and ultracentrifugation for sample preparation, these are excellent tools that analytical chemists should not neglect.
Another key advantage of removal of the nonpolars by precipitation is that these components tend to be highly retentive in reversed-phase LC, which can pose a serious problem with matrix effects and ghost peaks. Michlig and others studied this aspect in the analysis of highly complex hemp matrices (15). The precipitation cleanup greatly decreased the extent of matrix components coeluting with analytes as the mobile phase became less aqueous. Ghost peaks were also reduced by this type of cleanup, and the elimination of the possibility of ghost peaks was achieved by backflushing the columns with an organic solvent phase between injections (15). In long sequences, ghost peaks are often the cause of matrix effects in subsequent injections, despite the fact that cleanup may appear to be sufficient for individual samples. We have recently implemented alternating dual-column switching with backflushing in our QuEChERSER studies, which will be published in the future.
Salt-Out Partitioning and Automated µ-SPE Cleanup for GC Analysis: One of the elegant aspects of QuEChERSER is that the salt-partitioning and cleanup steps are tailored for GC-amenable analytes without compromise to accommodate LC analysis. After the small aliquot of initial extract is taken for LC, the remaining supernatant extract (10–11.7 mL) is decanted into 2 g of the same salt combination as used in QuEChERS. As demonstrated in the seminal publication (2), the use of MgSO4 combined (or not) with other salts for phase separation in QuEChERS is very robust in that different amounts of the salts per extract volume yield 100% recoveries for a wide range of GC-amenable pesticides. Thus, 2 g of the commercially available QuEChERS salts conveniently packaged in 15 mL centrifuge tubes were chosen for use in QuEChERSER. The protocol simply calls for pouring the supernatant (extract) into the tube containing the salts followed by 1 min shaking and 3 min centrifugation.
In the final step, automated instrument‑top sample preparation (ITSP or µ-SPE) is used for extensive cleanup (12–16,20,25–27). The approach is commercially available from several vendors for installation on any brand of GC. The analyst merely needs to transfer 1–1.5 mL of the acetonitrile extracts (upper layers) into autosampler vials and load the trays as normally done for instrumental sequences. The robotic liquid handler that also serves as an autosampler conducts the mini‑column SPE cleanup step for each extract just before GC injection. The cleanup is done in parallel with analysis of the previous sample, which is also done in parallel with LC analysis, to maximize sample throughput.
The same ITSP cleanup sorbent combination of 45 mg 20:12:12:1 and MgSO4–PSA–C18–CarbonX per 300 µL extract can be used in QuEChERS or QuEChERSER, but the fourfold less equivalent sample concentration in the latter method is much less likely to overload the mini-cartridges. Modern instruments are still capable of achieving the needed detection limits in QuEChERSER, and injection volume can be increased if needed.
The same sample–sorbent ratios could be conducted in batch-wise fashion using traditional cartridges loaded in a manifold or in a centrifugal SPE format, but this has at least four drawbacks compared to the automated approach: 1) traditional SPE costs more and involves attentive labour; 2) ITSP in parallel with analysis has higher sample throughput and permits larger batch sizes; 3) ITSP produces highly consistent, controlled, and slow (2 µL/s) flow rate for better precision; and 4) operation in batches loses the benefits of “just‑in‑time” cleanup prior to injection. The capital expense and maintenance of the robotic liquid handler are drawbacks with any automated system, but they should pay off in the long run.
In QuEChERS, d-SPE has several beneficial features (fast, flexible, easy, and inexpensive), but it sacrifices a degree of cleanup compared with column SPE. Automated µ-SPE has a similar extent of cleanup as cartridge-SPE, but it also possesses the advantages of speed and ease akin to d-SPE for reasonable cost and robustness. Although d-SPE has practical advantages over traditional SPE, it still takes time and care in pipetting to avoid sorbent particles from being transferred to the autosampler vials. QuEChERSER obviates this problem, and more importantly, it involves plenty of extract volume for easy and accurate transfers for both LC and GC analyses. A limited amount of final extract constitutes a drawback in QuEChERS, especially for reduced test portion size as in QuEChERSER.
Not only does QuEChERSER with ITSP recover a wider scope of analytes than QuEChERS with d-SPE, it also works without modification for a greater range of sample matrices. The effect of the increased solvent–sample ratio has already been discussed, but the more dilute extracts using the combination of cleanup sorbents in ITSP does an excellent job to remove common matrix components in meats (13), fish (14,15), hemp (16), eggs, grains, fruits, and vegetables (12). Although milk and soils have yet to be tested in QuEChERSER, the method is expected to work better than QuEChERS for those and other sample types. If needed, the same cleanup options can be done in QuEChERSER as in QuEChERS, such as hexane partitioning (21), enhanced‑matrix removal (28), magnetic‑SPE (29), and freezing out (6–9).
Analytical Aspects: Although analysis is distinct from sample preparation, the author would be remiss not to mention that low-pressure (LP) GC–MS (30) and analyte protectants (31) serve as valuable techniques in conjunction with QuEChERS, QuEChERSER, and other methods. LPGC matches the speed of ultrahigh-pressure liquid chromatography (UHPLC) with ≈13 min cycle times (from injection-to‑injection), which is approximately threefold faster than conventional GC analyses that would normally limit sample throughput. With respect to QuEChERS and QuEChERSER, LPGC enables large‑volume injection of acetonitrile in a standard hot splitless injector (12–16,30), which is a key feature to further lower detection limits. Similarly, QuEChERS and QuEChERSER are well-suited for using analyte protectants, which tend to be relatively polar sugar derivatives, because they are soluble in acetonitrile final extracts. Analyte protectants fill active sites in the GC–MS system, which acts to reduce peak tailing, improve accuracy, enhance signal, decrease matrix effects, and increase ruggedness in high-throughput analyses (31).
Last, targeted analysis in both LPGC and LPLC permits the use of summation function peak integration to greatly reduce analyst time spent on data review and manual re-integrations (32). Summation integration is commonly used when an analyte consists of multiple or misshapened peaks because the default integrator fails in those (and other) situations. Default software integrators do not provide fully trustworthy chromatographic peak integrations in mega-method analyses, especially at low concentrations. Despite that matrix interferants rarely occur when using highly selective MS-based detection, one set of integrator settings for each analyte rarely integrates all the peaks reliably throughout a sequence. However, summation function integration does not fail because it simply entails setting of the start and stop times for each analyte peak before and after the known retention times, accounting for peak widths. If the peaks consistently fall within the start and stop times (and there is a chromatographic problem if they don’t), then the peaks will always be integrated from the baseline, usually better than the conventional integrator or human can do. Then, MS-based identification criteria, including signal or concentration thresholds, indicate if the targeted analyte is present or not (33).
Conclusions
Technology advances in analytical chemistry and real-world laboratory methods need to be updated periodically to incorporate new proven capabilities. The QuEChERS approach was developed to suit the analytical devices and instruments commercially available 20 years ago, and the time has come for an updated generic sample preparation approach that incorporates modern technology. To meet this need, the QuEChERSER mega-method has been introduced (12–16), and its features in comparison with QuEChERS were described in this article.
QuEChERSER relies on the same three trends in sample preparation that have been taking place for centuries: miniaturization, streamlining, and automation. Using improved devices and techniques for comminution enables acceptable reduction of test portions sizes from 10–15 g to 1–5 g. Extraction and cleanup better tailored for both LC and GC analyses expands the analytical scope, including suitability to more applications. Using automated ITSP improves cleanup and analytical accuracy while also increasing ease and sample throughput. Even though QuEChERSER is ostensibly a sample preparation method, it follows the QuEChERS method in that it is also a holistic approach for streamlined generic sample preparation suited for modern times.
Acknowledgements
The author thanks Nicolas Michlig, Alan Lightfield, Yelena Sapozhnikova, Sergio Monteiro, and Ederina Ninga for their contributions in the development of QuEChERSER, and representatives of ITSP Solutions, CTC Analytics, Gerstel, ThermoFisher, Restek, UCT, Inc., Agilent, Sciex, Advanced Materials Technology, and Shimadzu who were supportive and helpful in QuEChERSER studies. The author also thanks Doug Raynie and Laura Bush for the kind invitation to submit this article.
Disclaimer
The mention of a brand or firm name does not constitute an endorsement by the U.S. Department of Agriculture (USDA) above others of a similar nature not mentioned. USDA is an equal opportunity provider and employer.
References
- M. Anastassiades, S.J. Lehotay, and D. Štajnbaher, 18th Annual Waste Testing & Quality Symposium Proceedings, Arlington, Virginia (USA), pp. 231–241 (2002).
- M. Anastassiades, S.J. Lehotay, D. Štajnbaher, and F.J. Schenck, J. AOAC Int. 86, 412–431 (2003).
- S.J. Lehotay, J. AOAC Int. 90, 485–520 (2007).
- P. Payá, M. Anastassiades, D. Mack, I. Sigalova, B. Tasdelen, J. Oliva, and A. Barba, Anal. Bioanal. Chem. 389, 1697–1714 (2007).
- S.J. Lehotay, M. Anastassiades, and R.E. Majors, LCGC Europe 23, 418–429 (2010).
- R. Perestrelo, P. Silva, P. Porto-Figueira, J.A.M. Pereira, C. Silva, S. Medina, and J.S. Câmara, Anal. Chim. Acta 1070, 1–28 (2019).
- Á. Santana-Mayor, B. Socas-Rodríguez, A.V. Herrera-Herrera, and M.Á. Rodríguez-Delgado, J. Trends Anal. Chem. 116, 214–235 (2019).
- L. Kim, D. Lee, H.-K. Cho, and S.-D. Choi, Trends Environ. Anal. Chem. 22, e00063 (2019).
- S.J. Lehotay and Y. Chen, Anal. Bioanal. Chem. 410, 5331–5351 (2018).
- S.J. Lehotay and A.R. Lightfield, J. AOAC Int. 103, 584–606 (2020).
- B. Greer, O. Chevallier, B. Quinn, L.M. Botana, and C.T. Elliott, Trends Anal. Chem. 141, 116284 (2021).
- S.J. Lehotay, N. Michlig, and A.R. Lightfield, J. Agric. Food Chem. 68, 1468–1479 (2020).
- S.H. Monteiro, S.J. Lehotay, Y. Sapozhnikova, E. Ninga, and A.R. Lightfield, J. Agric. Food Chem. 69, 1159–1168 (2021).
- E. Ninga, Y. Sapozhnikova, S.J. Lehotay, A.R. Lightfield, and S.H. Monteiro, J. Agric. Food Chem. 69, 1169–1174 (2021).
- N. Michlig, S.J. Lehotay, A.R. Lightfield, H. Beldoménico, and M.R. Repetti, J. Chromatogr. A 1645, 462097 (2021).
- S.H. Monteiro, S.J. Lehotay, Y. Sapozhnikova, E. Ninga, G.C.R. Moura Andrade, and A.R. Lightfield, Food Addit. Contam. A (in press).
- M. Bruce and D.E. Raynie, LCGC Europe 30, 638–641 (2017).
- S.J. Lehotay and J.M. Cook, J. Agric. Food Chem. 63, 4395–4404 (2015).
- M. Roussev, S.J. Lehotay, and J. Pollaehne, J. Agric. Food Chem. 67, 9203–9209 (2019).
- S.J. Lehotay, L. Han, and Y. Sapozhnikova, Anal. Bioanal. Chem. 410, 5465–5479 (2018).
- S.J. Lehotay, A.R. Lightfield, L. Geis-Asteggiante, M.J. Schneider, T. Dutko, C. Ng, L. Bluhm, and K. Mastovska, Drug Test. Anal. 4(Suppl. 1), 75–90 (2012).
- USDA Food Safety and Inspection Service, Chemistry Laboratory Guidebook CLG-MRM1-3 (www.fsis.usda.gov/news-events/publications/chemistry-laboratory-guidebook), accessed December 2021.
- S.J. Lehotay and A.R. Lightfield, Anal. Bioanal. Chem. 413, 3223–3241 (2021).
- V. Joshi and W.K. Way, Am. Pharm. Rev. 23, 82–83 (2020).
- B.D. Morris and R.B. Schriner, J. Agric. Food Chem. 63, 5107–5119 (2015).
- S.J. Lehotay, L. Han, and Y. Sapozhnikova, Chromatographia 79, 1113–1130 (2016).
- E. Hakme and M.E. Poulsen, J. Chromatogr. A 1652, 462384 (2021).
- T. Anumol, S.J. Lehotay, J. Stevens, and J. Zweigenbaum, Anal. Bioanal. Chem. 409, 2639–2653 (2017).
- H.-L. Jiang, N. Li, L. Cui, X. Wang, and R.-S. Zhao, Trends Anal. Chem. 120, 115632 (2019).
- S.J. Lehotay, J. de Zeeuw, Y. Sapozhnikova, N. Michlig, J. Rousova Hepner, and J.D. Konschnik, LCGC N. Am. 38, 457–466 (2020).
- R. Rodríguez-Ramos, S.J. Lehotay, N. Michlig, B. Socas-Rodríguez, and M.Á. Rodríguez-Delgado, J. Chromatogr. A 1632, 461596 (2020).
- S.J. Lehotay, LCGC Europe 30, 530–540 (2017).
- S.J. Lehotay, Anal. Bioanal. Chem. (in press) (doi: 10.1007/s00216-021-03380-x)
About The Author
Steven Lehotay is Lead Scientist in the US Department of Agriculture’s Agricultural Research Service at the Eastern Regional Research Center in Wyndmoor, Pennsylvania, USA.
About The Column Editor
Douglas E. Raynie is Department Head and Associate Professor at South Dakota State University, USA. His research interests include green chemistry, alternative solvents, sample preparation, high-resolution chromatography, and bioprocessing in supercritical fluids. He earned his Ph.D. in 1990 at Brigham Young University. Raynie is a member of LCGC’s EAB. Direct correspondence about this column via e-mail toamatheson@mjhlifesciences.com












This information is supplementary to the article “Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing”, which was published in the May 2025 issue of Current Trends in Mass Spectrometry.


Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).

Evaluating Natural Preservatives for Meat Products with Gas and Liquid Chromatography
April 1st 2025A study in Food Science & Nutrition evaluated the antioxidant and preservative effects of Epilobium angustifolium extract on beef burgers, finding that the extract influenced physicochemical properties, color stability, and lipid oxidation, with higher concentrations showing a prooxidant effect.


