Supercritical Fluid Chromatography for Chiral Analysis, Part 2: Applications
In the second part of this review article, the recent progress in supercritical fluid chromatography (SFC) for enantiomeric separations is evaluated. With the substantial developments carried out over the past years in instrumentation, columns, and detector hyphenation, the interest in chiral SFC has been steadily growing in various fields. In combination with novel developments in chiral stationary phase chemistries, the enantioselective analysis range has been significantly extended. Several applications reported on the enantioselective separation of drugs and pharmaceutical compounds using chiral SFC are discussed, including pharmaceutical applications, clinical research, forensic toxicology, and environmental sciences.



Over the past decade, particularly since the commercialization of the first high-pressure and low-dispersion instruments, supercritical fluid chromatography (SFC) has become a more established technique.
Since the introduction of polar co-solvents as a mobile phase, SFC has been applied to various fields, ranging from fossil fuels and polymers to food and pharmaceutical applications. Chirality also plays a significant role in these research areas, as enantioselective separations are often required to discriminate between enantiomers showing different pharmacological or biological activities (1). Based on the advantages listed in part 1 of our review (2), it does not come as a surprise that an increased number of studies using SFC for chiral applications have been reported in the literature.
The instrumental improvements, such as pressure stability and UV-noise reduction, but also the commercialization of novel polar and nonpolar stationary phases, coated or immobilized on to smaller and superficially porous particles, have led to increased performance in chiral SFC analysis (3). These developments provide a powerful orthogonal and versatile analytical approach for high-efficient and high-throughput chiral assessments. Moreover, the developments in SFC–mass spectrometry (MS) have also strengthened the position of chiral SFC, especially when a certain level of sensitivity or additional selectivity is desired. In the past decade, SFC has been increasingly used for chiral analysis—most notably in the field of pharmaceutical analysis, clinical applications, forensic toxicology, and environmental analysis, as discussed in the following sections. The most significant requirements in those areas are speed (pharmaceutical), sensitivity (clinical, forensic toxicology, and environmental analysis), and selectivity (illicit drug analysis). The aforementioned unique properties of supercritical fluids and recent innovations in SFC can make an important contribution to tackling these challenges.


Chiral SFC in Pharmaceutical Analysis
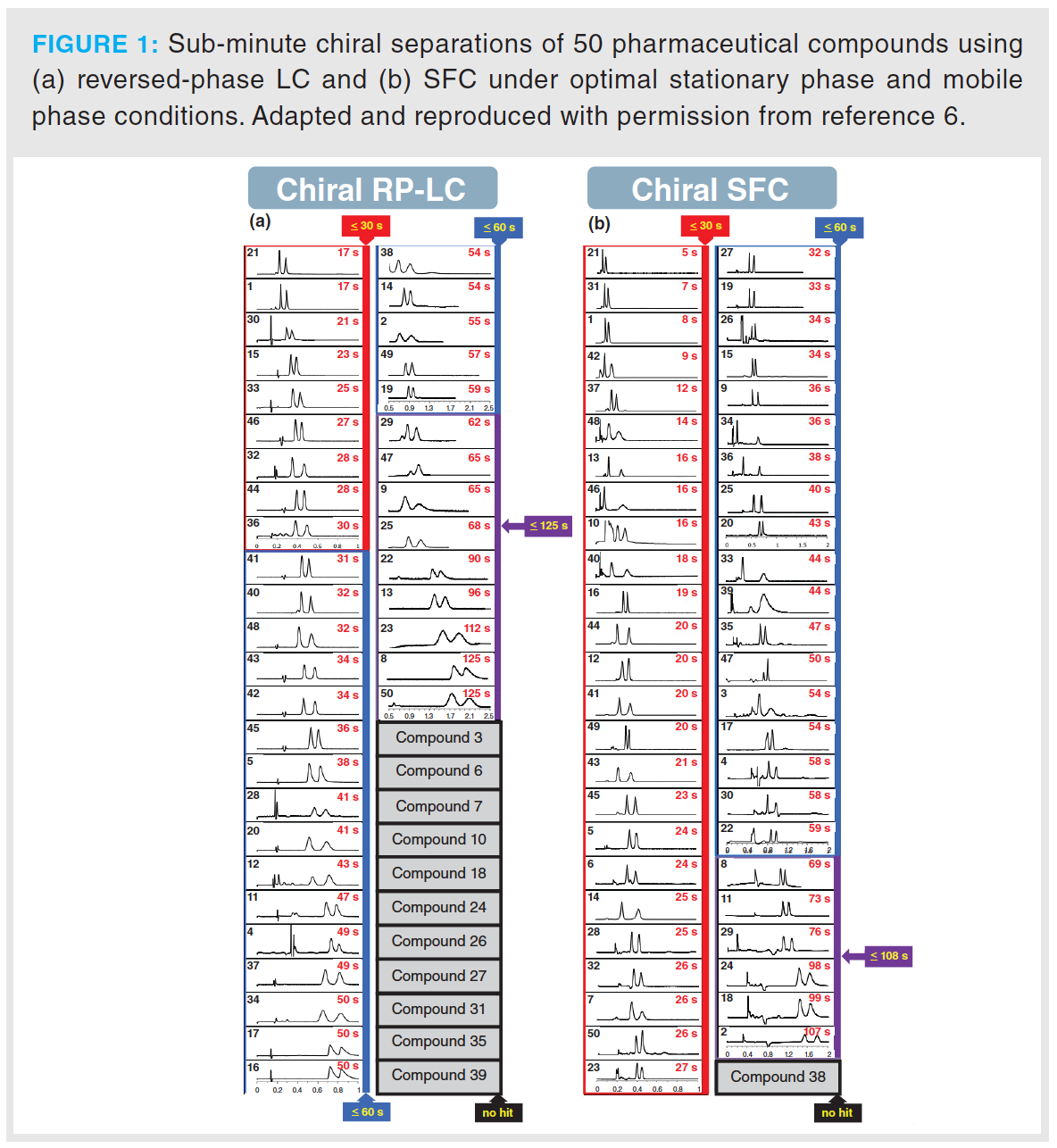
Due to the expected differences in pharmacological activity of enantiomers, the spatial configurations of new pharmaceutical compounds need to be thoroughly investigated in the drug development pipeline. Table 1 lists an overview of the pharmaceutical applications reported since 2015,
where SFC has been used for enantioselective separations (4,5). Similar to chiral liquid chromatography (LC)-based applications, polysaccharide-based chiral stationary phases (CSPs) have been most frequently employed. The broad applicability and high-loadability of these phases support their wide use in drug development. Only a few studies reported optimal separation using macrocyclic glycopeptides, cyclofructan-based, and Pirkle-type CSPs. Other types of CSPs, such as polymer- and cyclodextrin‑based, have not been found in recent publications. In this field of application, 3- and 5-μm fully porous particles (FPP) are the most common particles sizes for cellulose- and amylose-based columns. Smaller fully and superficially porous particles (SPP) are less common, but have proved to be successful in combination with macrocyclic glycopeptide- and cyclofructan-based stationary phases (6–8).

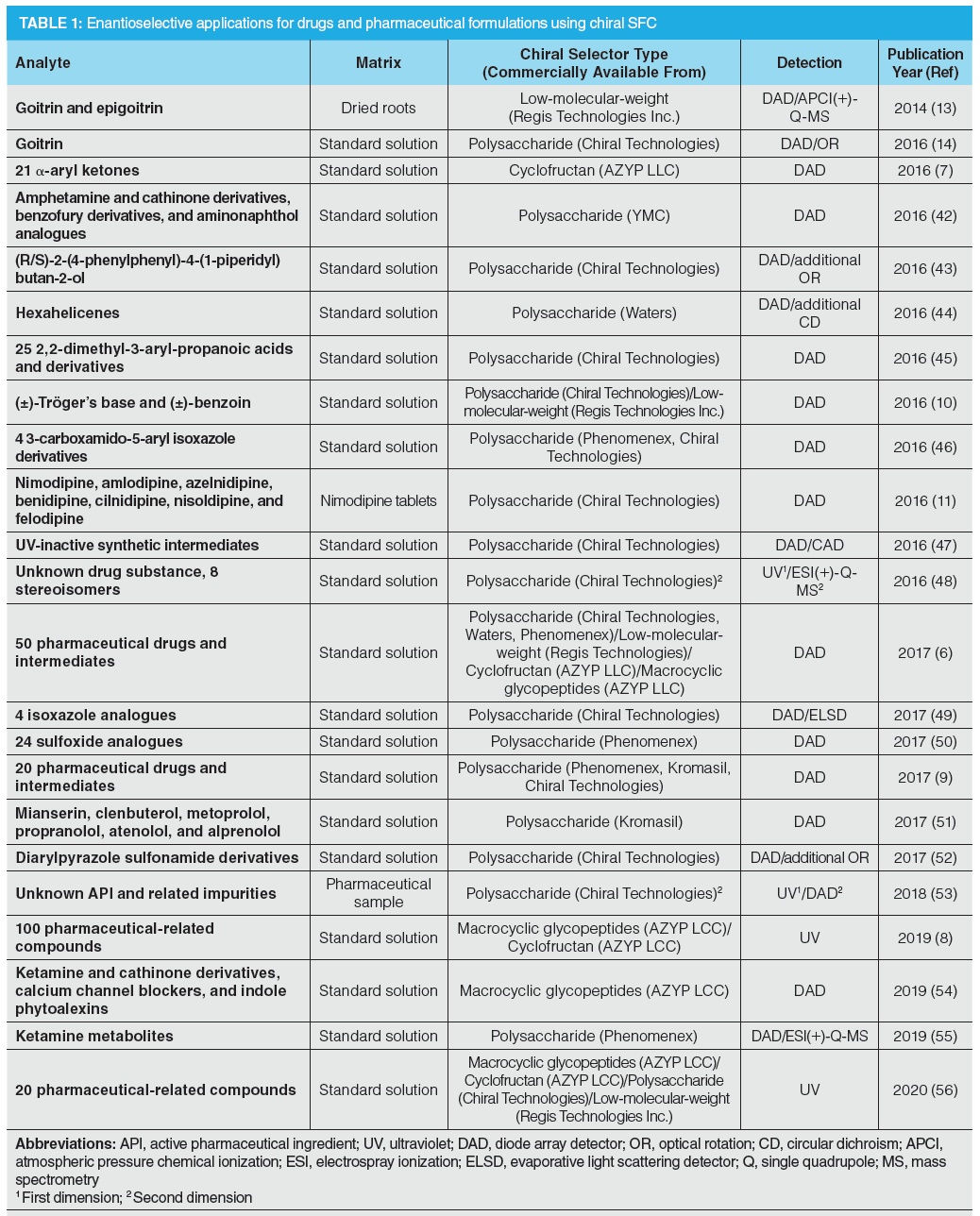
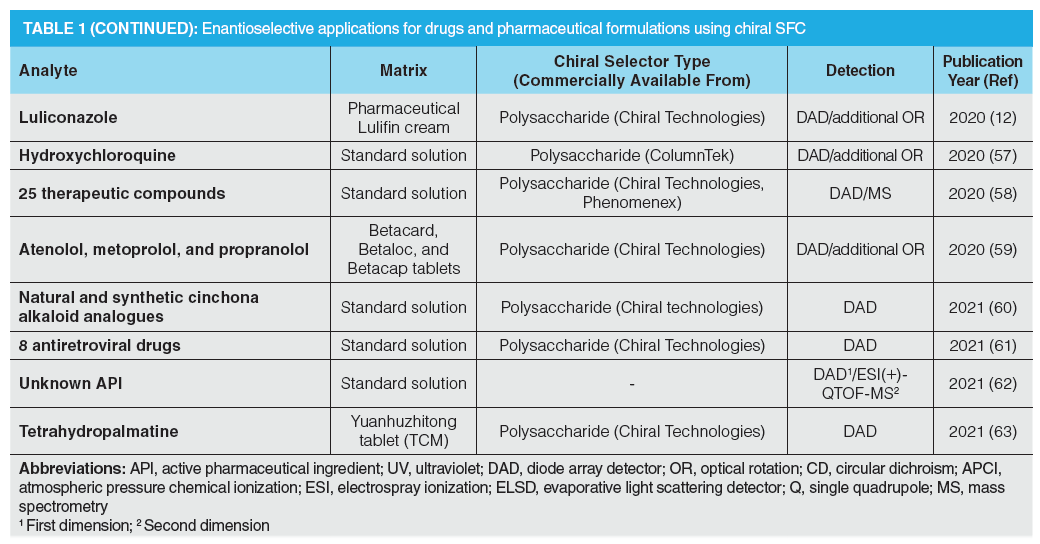
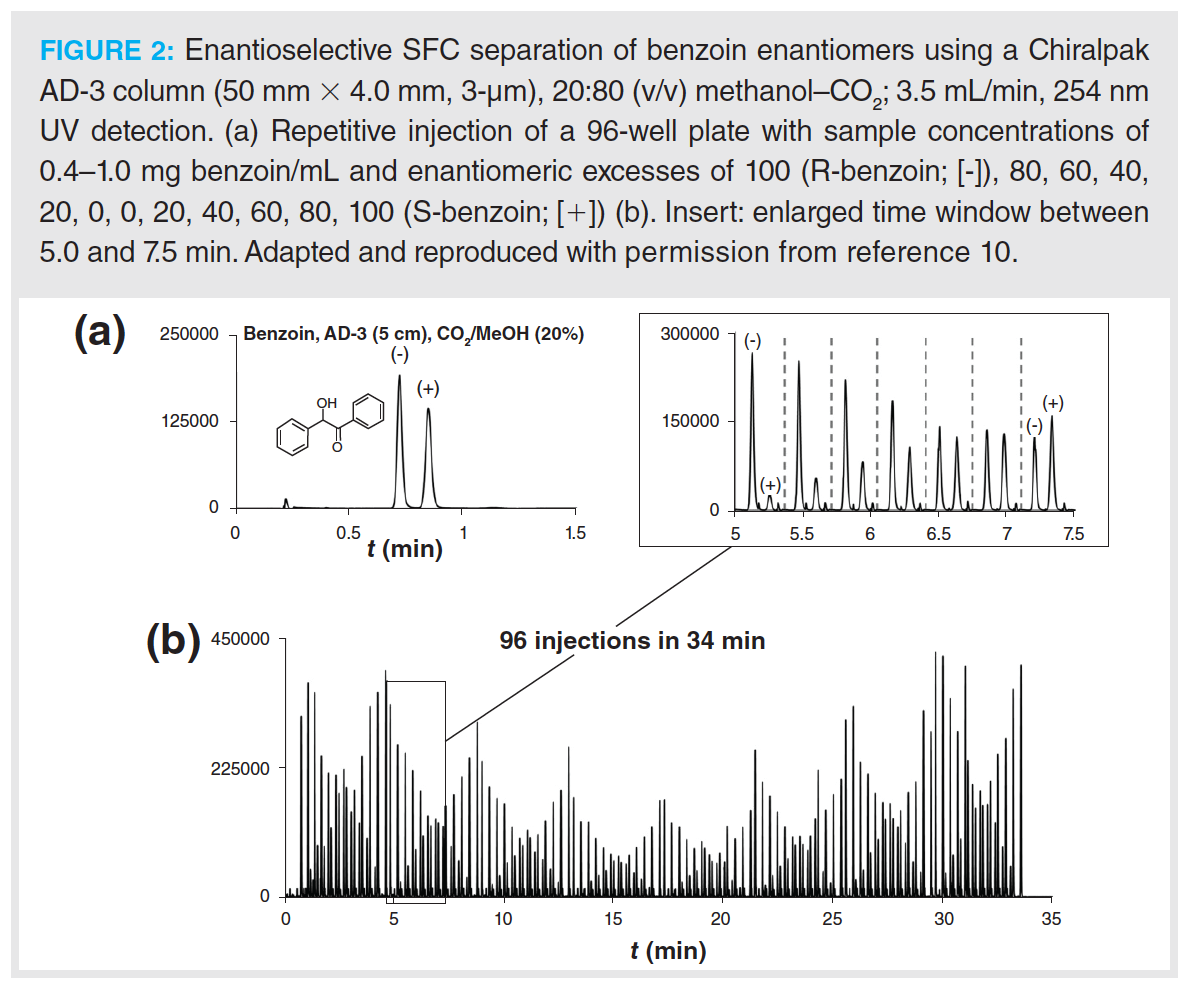
In the pharmaceutical industry, high‑throughput approaches are required during purity assessment tests. Due to the unique features of supercritical fluids, higher flow rates can be applied in SFC, which allow for high-throughput analysis. Barhate et al. showed that sub-minute separations are achievable with SFC after the selected drugs undergo an extensive column screening, followed by a method optimization when necessary (6), as illustrated in Figure 1. The overall goal of the study was to determine the feasibility of ultra-fast chromatographic chiral separations using improved CSPs, instrumentation, and particle technology. Nováková and Douša developed a new rapid and general screening and optimization approach for enantioselective SFC that is based on the enantioseparation success rates of different parameter settings (9). Rapid separations are helpful for the implementation of new high-throughput strategies, such as the so-called multiple injections in a single experimental run (MISER) to reduce analysis time and simplify data analysis (10). This approach performs rapid-fire high-throughput injections of multiple samples, resulting in multiple sub-minute (enantioselective) separations within a single chromatographic run. Figure 2 presents a flow injection analysis-like chromatogram of benzoin enantiomers (10).

Zhang et al. demonstrated the enantioselective analysis of various 1,4-dihydropyridines in a pharmaceutical formulation using chiral SFC and immobilized polysaccharide-based CSPs (11). Using a miniaturized sample preparation approach, that is, a two‑phase hollow fibre-based liquid‑phase microextraction (2p-HF‑LPME), the concentrations of nimodipine enantiomers in tablets were determined, with recoveries up to 99.8%. Pandya et al. showed the successful separation of four stereoisomers of luliconazole in a cream formulation (12). In addition, they determined the relation between elution order and binding energies, enabling a better understanding of the different interactions involved during the chiral recognition of luliconazole stereoisomers. The main reason for choosing chiral SFC over chiral LC was the speed of analysis, with the added advantage of minimizing toxic organic waste generation.
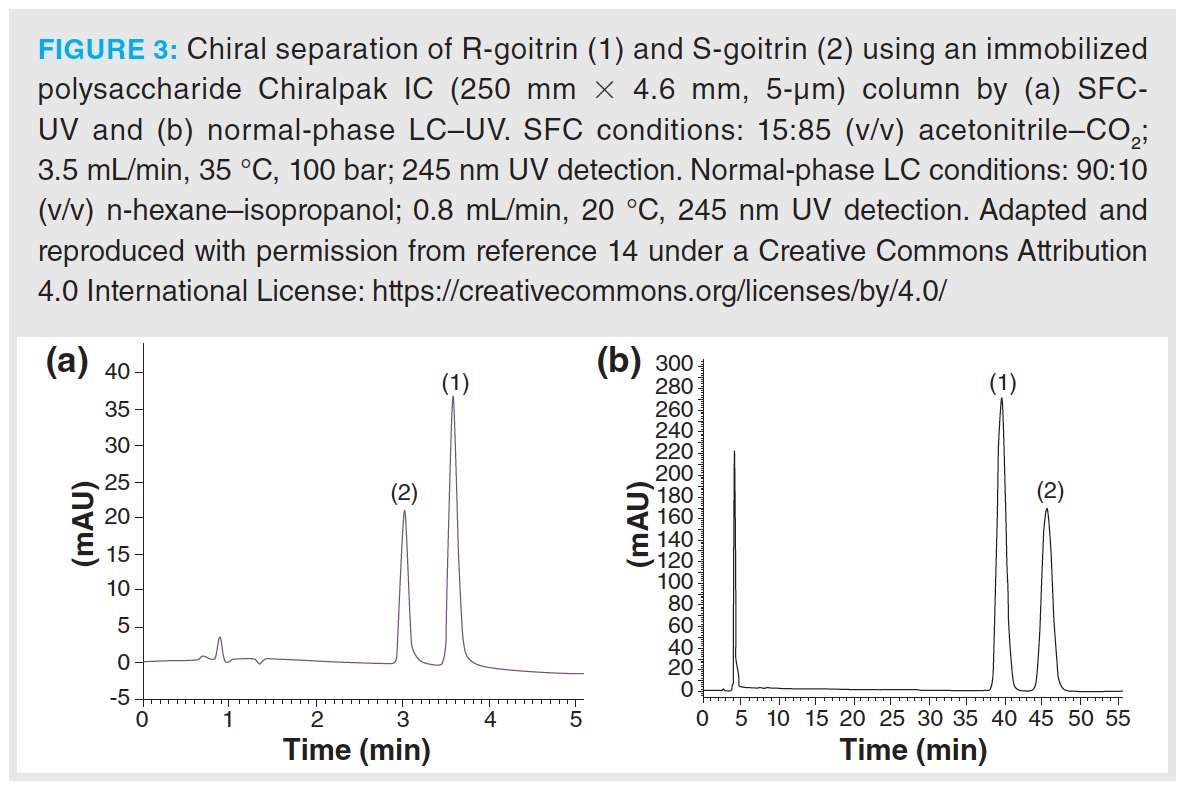
Traditional Chinese drugs (TCD) also contain pharmaceutically active compounds with one or multiple chiral centres. An example is the dried roots of Isatis Indigotica Fort, which is traditionally used against fevers, and contains the substance goitrin. (R)-goitrin is considered the therapeutic form, while its enantiomeric pair causes adverse effects on the thyroid gland (13). Wang et al. developed an SFC-photo diode array-MS (SFC–PDA–MS) approach to enable the analysis of the enantiomeric forms of goitrin in both dried root extracts and commercially available pharmaceutical formulations.
The proposed method, based on a Pirkle-type CSP, led to the successful separation from plant matrix interferences, while it simultaneously provided enantiomeric separation. The developed method was eight times faster than the traditional normal-phase LC–UV analysis but with lower sensitivity. Nie et al. later developed an analytical and preparative method for the analysis of the same drug, using an immobilized cellulose-based CSP that led to improved chromatographic efficiency and analysis time. The comparison of both methods is shown in Figure 3 (14).

Clinical Applications
Once a drug is administered, chiral inversions may change the spatial configuration of a drug molecule and its metabolites (15,16). These in vivo by‑products and intermediates should thus be monitored and individually evaluated for their pharmacologic activities. Biofluids, such as plasma and serum, are complex matrices, with thousands of endogenous analytes that can affect the analytical performance. Moreover, a parent drug and its potential metabolites are typically present at low concentration levels. In this context, SFC–MS is often required to achieve sufficient selectivity and sensitivity for compound identification and quantification. Table 2 provides an overview of the clinical applications reported for the analysis of drugs and drug metabolites since 2015 (4,5).

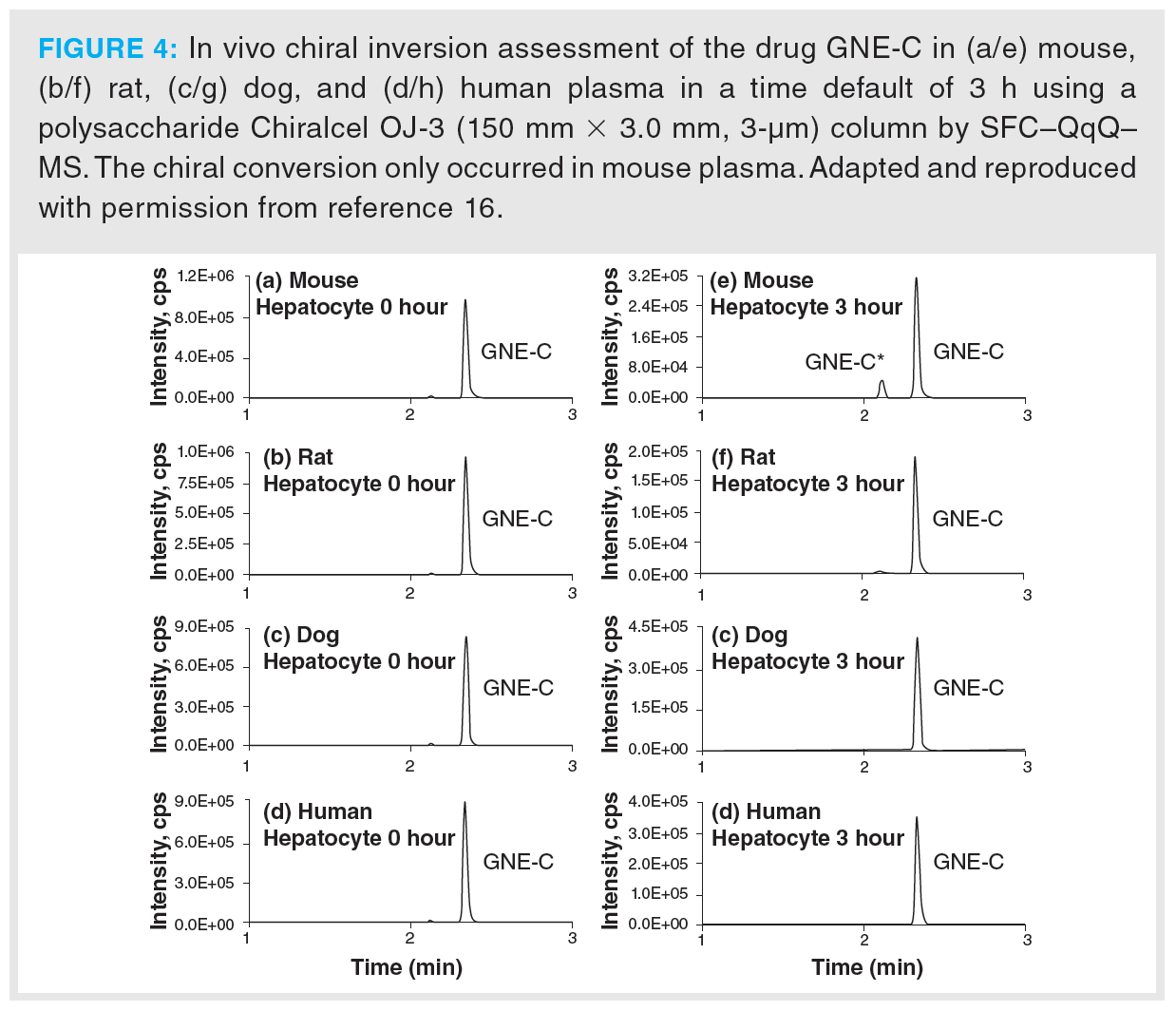
A study by Chen et al. showed the applicability of SFC–MS/MS to investigate the potential in vivo chiral inversion of three different novel drugs in various mammal plasmas (16). While the drugs GNE-A and GNE-B showed no inversion a few hours after administration, inversion occurred with GNE-C up to 25%. However, as is shown in Figure 4, inversion was only observed in mouse plasma. For that reason, chiral inversions do not only require a specific chiral centre, they also depend on the biological environment in which the chiral drug is located. A study by Licea-Perez et al. demonstrated the chiral inversion of the novel (S)-GSK-B drug to (R)-GSK-B (17). While the method was successful for the analysis of standards, no inversion was detected in rat plasma after administration.

Su et al. employed SFC–MS/MS to study the pharmacokinetics of the chiral drug rabeprazole (18). After administration of racemic rabeprazole to beagle dogs, repetitive chiral analysis of their serum was performed, showing a change in the enantiomeric ratio over time. This shift indicated that rabeprazole exhibited stereoselective pharmacokinetics properties. One of these enantiomers was then more effective than the other.
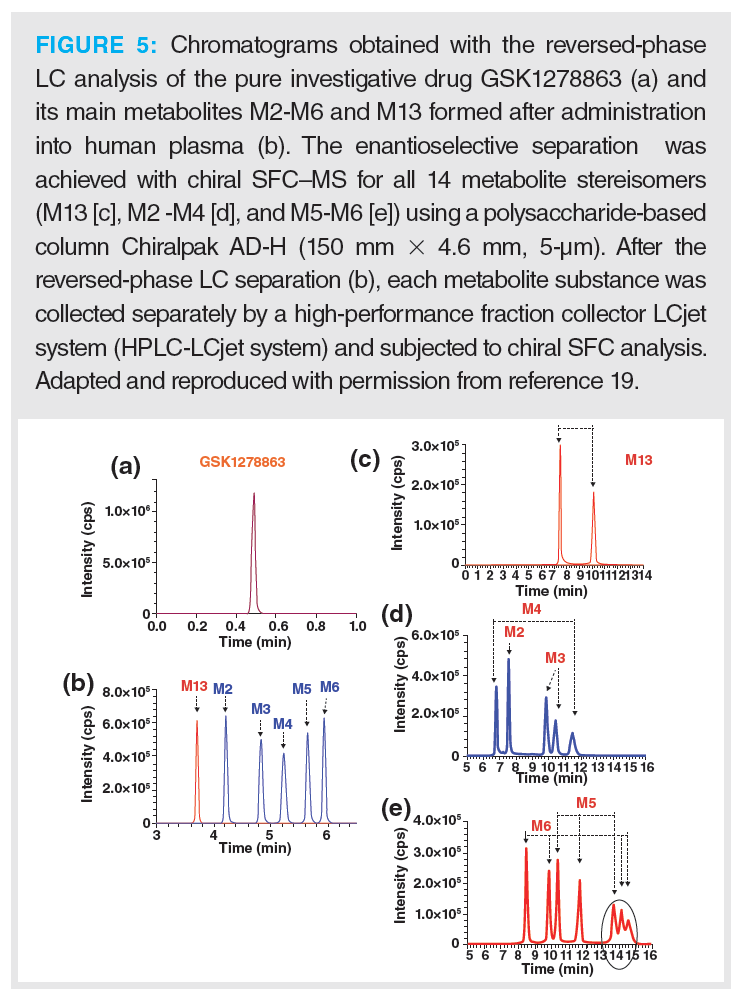
Drug metabolites are also relevant, as they can be either chiral or achiral. After detecting these minor by-products in vivo, the potential presence of their stereoisomers must be monitored and their pharmacological activities assessed. Another study by the group of Licea-Perez investigated the drug metabolism of the drug GSK1278863, developed for the treatment of anaemia correlated to chronic kidney disease (19). An SFC–MS/MS method was developed for the detection of six predominant metabolites formed by cytochrome P450 enzymes, which are responsible for the formation of 14 stereoisomers. Figure 5 shows the stepwise analyzing process for the full (enantiomeric) assessment of this investigative drug (19).

Online supercritical fluid extraction and chiral SFC–MS (SFE–SFC–MS) analysis have been performed by Hofstetter et al. for the extraction and quantification of ketamine enantiomers and their chiral metabolites in a dried urine sample (20). It proved to be the most efficient approach for ketamine analysis and was the first SFE–SFC–MS method to be validated according to the international EMA regulatory guidelines. Automated SFE was a more time-efficient approach to extract the relevant ketamine substances than via the previously described liquid–liquid extraction (LLE) method; it also reduced the necessary amount of biological matrix (20 μL) required for enantioselective quantification (21). The use of SFE–SFC demonstrated promising possibilities and represents an interesting approach in the pharmaceutical industry.
Forensic Toxicology
Chiral SFC has been used in doping research to determine whether athletes have been exposed to a doping agent by drug administration or by consumption of specific foods. An example was presented by Parr et al. where SFC–MS/MS was used to trace the origin of the doping agent clenbuterol by determining the enantiomeric ratio (22). Previous studies showed that the concentration of clenbuterol enantiomers in meat changed due to (different) pharmacological activities in animals, while no changes were observed in human urine within 18 h of administration. This difference in enantiomeric ratio can therefore provide information about the origin of the doping agent and ensure that athletes can be fairly judged.
Enantioselective SFC has been increasingly used in forensic investigations for a variety of illicit and licit drug-related purposes (3). Novel psychoactive substances (NPS) may be one of the most important motivators for chiral studies of these new derivatives (23). Due to their novelty, little is known about the pharmacological activities of NPS and their enantiomeric counterparts. Enantioselective separations are therefore of great importance to help identify NPS and evaluate their effects on the human body. Additionally, chiral assessments can provide information about the time of consumption by monitoring the enantiomeric ratio in human serum, as demonstrated by Losacker et al. using LC–MS/MS (24). However, this latter application has not yet been described for chiral SFC. Table 3 lists an overview of various forensic toxicology applications from the past five years using chiral SFC (4).

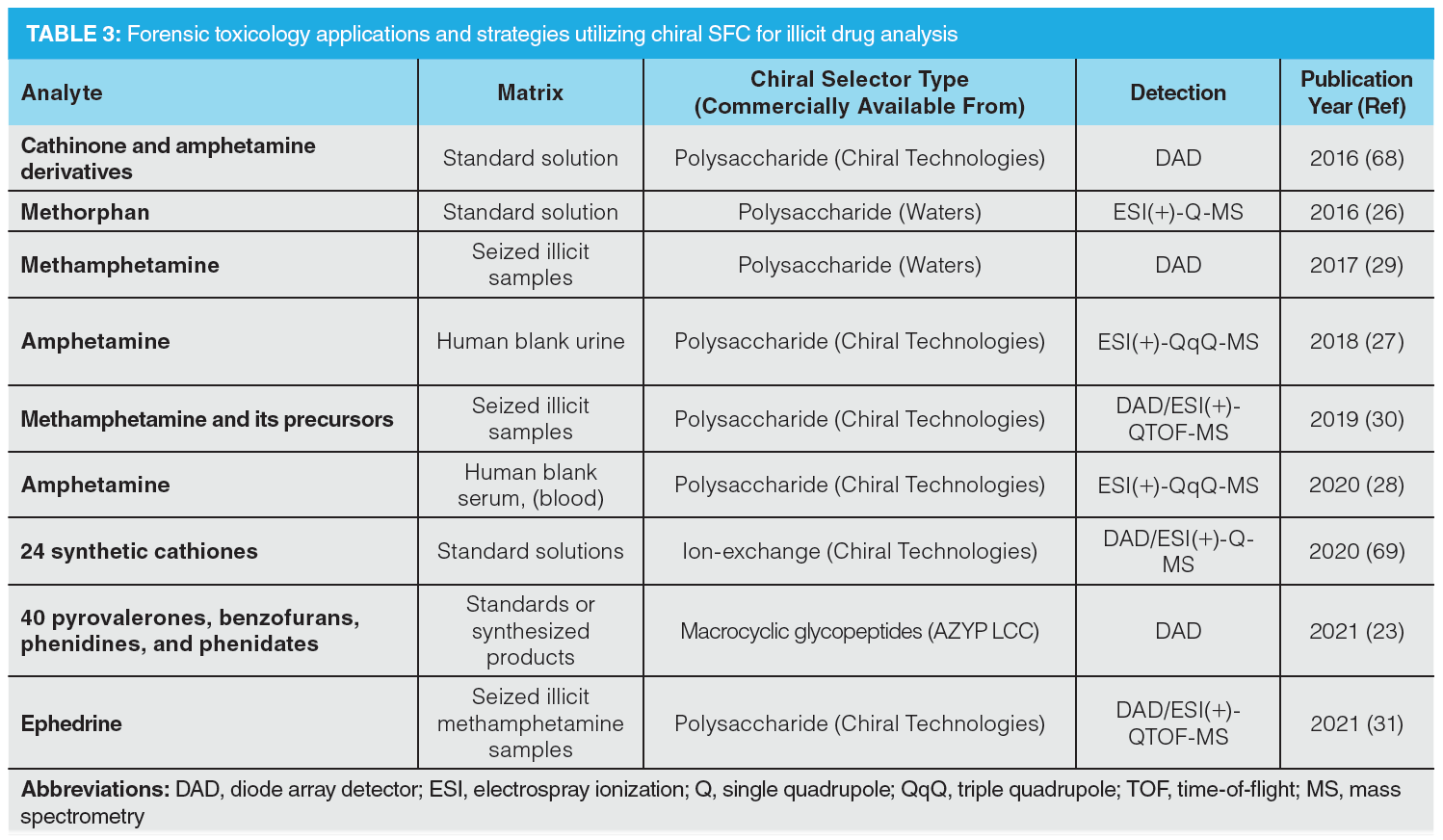
In a recent work by Folprechtová et al., various NPS classes (pyrovalerone, benzofuran, phenidine, and phenidate derivatives) were analyzed by SFC and LC using a core–shell vancomycin‑based CSP (23). The vancomycin-based chiral selector showed high enantioselective properties for both approaches, resulting in fast and highly efficient enantiomeric separations. This study showed the complementarity between SFC and LC for chiral analysis. Overall, 88% of the NPS enantiomers were separated by SFC, while only 69% of NPS enantiomers were separated with LC. Both approaches were potentially able to separate most of the known NPS enantiomers, as well as those yet to be discovered. Additionally, the vancomycin-based methods can be used for the analysis of seized samples and perform simultaneous multiple enantiomeric separations—beneficial for full purity profiling.
Some listed drugs are not only used for recreational purposes but are also administered for medical reasons. It is therefore of great importance to be able to distinguish between these two scenarios of administration for lawful conviction. Dextromethorphan (R-enantiomer), for example, is an antitussive drug that can be purchased without a prescription. Levomethorphan (S-enantiomer), however, is a strong narcotic analgesic and is registered as a controlled substances in many countries (25). Li et al. developed a direct enantioselective SFC–MS method to separate these enantiomers within 3 min using an amylose-based CSP (26). A similar study was performed by Hegstad et al., in which an SFC–MS/MS method was developed to separate and quantify (R/S)-amphetamines in urine, to distinguish between medical (legal) and recreational (illegal) intake (27). In most cases, the pure S-enantiomer of amphetamine or its prodrug was prescribed as the medical drug, while illicit amphetamine mainly occured as a racemic mixture. The developed SFC approach has recently been extended by Havnen et al. to analyze serum and blood samples (28).
Segawa et al. demonstrated in successive studies the potential of using those particular enantiomeric ratios to trace back the origin of seized methamphetamine by identifying their precursors and synthetic route (29,30,31). Together with various ephedrine derivatives (EP derivatives) and phenyl-2-propanone (P2P) as starting material, and different optional reaction pathways, there are many ways to produce methamphetamine (MA; crystal meth). Segawa et al. demonstrated a simultaneous chiral analysis of methamphetamine and its precursors using a polysaccharide‑based CSP analyzed with SFC–MS/MS (30). Such an approach allows for a direct and fast determination of a potential synthetic route. Additionally, each of these synthetic routes leads to their own typical chiral MA impurities in specific enantiomer ratios, which makes it possible to trace back the original precursor and method used. This information can be helpful by linking the seized drugs to potential clandestine production laboratories.
Segawa et al. extended this chiral SFC profiling approach to the analysis of seized MA by including the analysis of EP stereoisomers (31). It has recently been reported that ephedrine and pseudoephedrine are by-products from the P2P synthesis. Both forms of ephedrine are therefore ultimately present in the phenyl-2-propnanone-based MA samples and can be used for sample identification.
Environmental Studies
Pharmacological active compounds (PACs) are increasingly seen as major pollutants within ecological systems. However, despite the known concerns of these drug stereoisomers, research on their ecotoxicological effect is still limited (32). One of the reasons for this shortfall is the lack of knowledge on the potency of different enantiomeric forms, as well as sensitive and efficient methods. Besides in vivo inversions and metabolism after administration, drug substances may undergo further modifications initiated by wastewater treatments and biological processes (33). These reactions are usually enantioselective and therefore enrich or deplete one enantiomeric form over the other, which may increase the environmental impact. Table 4 lists the various applications in which chiral SFC has been implemented into environmental studies (4,5).


Sensitive chiral systems are essential to detect even the most diluted substances as well as their enantiomeric opposites to fully profile the drug content in water. Rice et al. developed an SFC–MS/MS method for multiresidue analysis of both chiral and achiral PACs in waste- and natural waters (32). Over 80 PACs were successfully separated within 10 min of analysis, including enantiomeric separations using a polysaccharide-based CSP. Additional chiral analysis proved the occurrence of configurational modifications, as the uneven enantiomeric ratios in influent, effluent, and natural water differ from each other. However, as these water samples were collected at one specific location and time point, no firm conclusions could be drawn.
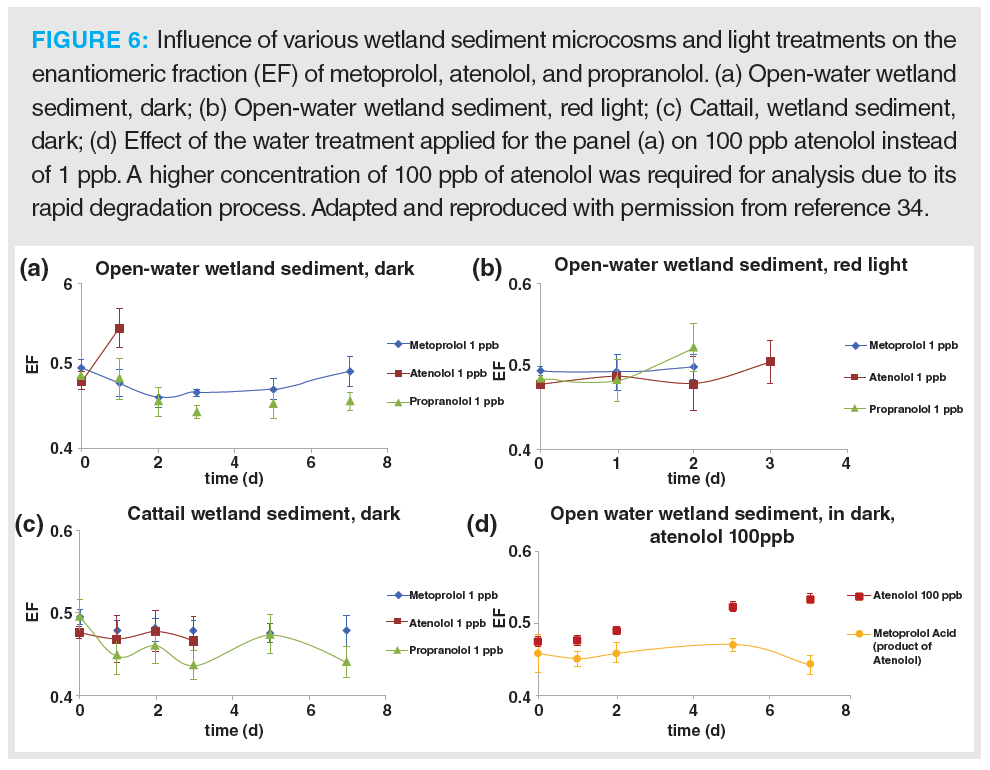
Racemic drugs are most likely affected by in vivo and/or natural microbiological reactions. Earlier studies by Svan et al. and Camacho-Muñoz et al. using (UHP)SFC–MS/MS and polysaccharide‑based CSPs showed similar activities in enantiomeric conversions of various PACs in wastewater and wetland microcosm samples (34,35). Figure 6 shows the influence of different wetland sediment environments on the enantiomeric fraction of metoprolol, atenolol, and propranolol (34). No significant changes occurred at 1 ppb, but atenolol showed a slight increase at 100 ppb. This indicates an enantioselective enzymatic process that may participate in its breakdown.

SFC proved to be a worthy alternative for efficient analysis of environmental samples, by allowing for enantiomer‑specific evaluations on the ecological impact of drugs. Analysis of influent and effluent water can also provide additional information about the use of drugs within a specific area, including whether it is legal (prescribed) and/or illegal (recreational) consumption. Finally, this information may be used in pharmaceutical studies to complement in vivo metabolomics-based approaches or within forensic investigations for determining the origin of the drugs.
Discussion and Conclusion
The technological developments in (chiral) SFC have made this a competitive technique for enantioselective separations with respect to conventional LC. Advantages include the achievement of efficient separations within a reduced analysis time and the replacement of toxic solvents (for example, from reversed-phase LC) with CO2. These have been covered in more details in part 1 of this review (2).
In chiral analysis, method optimization remains a complex and timely process. Notably, the selection of the adequate CSP column is a time-consuming but essential screening step, since each chiral selector interacts differently with a specific chiral molecule. Hundreds of CSPs have been developed, without one specific phase that can be labelled as universal. Such a CSP would be highly beneficial, as this would significantly simplify the method development. Future studies focusing on the development of universal enantioselective column chemistries are therefore needed to ensure optimal chiral distinctions for as many different stereoisomers as possible. In parallel, innovative workflows enabling the screening of a large diversity of phases are also beneficial. These workflows can be specialized for single column systems (36,37,38), tandem column systems (39,40), or stereoisomers with multiple chiral centres (MCC) (41). De Klerck et al. proposed a generic screening strategy when investigating the effect of different CSPs in combination with varying mobile-phase compositions, and used principle component analysis (PCA) to confirm selectivity differences between the screened SFC systems (36). Hegade et al. proposed a new effective screening method called stationary-phase optimized selectivity supercritical fluid chromatography for the prediction of elution profiles of tandem column systems (40). This approach is based on the combined interaction models from the individual columns used. Lin et al. developed an effective screening strategy for stereoisomers with single and multiple chiral centres, answering several key questions regarding which parameters to use for optimal MCC compound coverage (41).
The potential of chiral SFC application outside of the pharmaceutical industry has been minimally explored so far. A few publications have demonstrated promising applications and strategies but only for a limited number of drugs. Further implementation of chiral SFC into clinical, forensic, and environmental drug‑related studies are therefore encouraged to foster the acceptance of chiral SFC in a broad spectrum of real-life cases. Specifically, the determination of the ecotoxicological effects of drugs after consumption deserves more attention and should be more predominant within drug development processes. In addition, successive studies can be of added value to determine how the obtained data can be used in a larger perspective. Further implementation and new developments are expected to be promising based on the cost-saving and green advantages, applications, and strategies mentioned in this review.
SFC mechanisms exhibit high orthogonality compared to LC-based separation techniques. With the development of special interfaces, the rise of multidimensional applications with SFC included as one of the two dimensions is, therefore, no surprise. Various setups for chiral separations have been proposed, demonstrating a highly selective and efficient achiral impurity assessment in the first dimension and chiral distinction in the second. Despite the good enantiomeric resolution acquired, sensitivity in 2D applications remains a challenge because it is important to detect trace-level impurities. Moreover, hardware and solvent incompatibility may form a limitation in this field of application. Further studies and instrumental developments are required to tackle these drawbacks and would be of great value to fully assess (enantiomeric) purity or activity within a single run. Still, the complexity and input required to fully optimize such a system for integrated achiral and chiral impurity assessment is intensive and time-consuming. It is therefore recommended to first apply simpler optimization approaches to resolve difficult enantiomers rather than directly developing a 2D-system. Such approaches include changing the CSPs, tandem column setups, or selecting a different detector. Nevertheless, further studies in 2D chiral applications are still important because this approach may be required in complex cases.
Nowadays, chiral SFC is not only recognized for its success on a preparative scale but has also been demonstrated to be a worthy asset for analytical purposes. SFC provides high-efficient and complementary chiral separations, allows high-throughput analysis, is eco-friendlier and safer than traditional chiral normal-phase LC, and is cost-efficient. Despite these advantages, there are still some challenging hurdles to overcome. Bottlenecks are undefined chiral recognition mechanisms, lack of highly universal CSPs, time-consuming screening approaches, relatively low maximum system back pressure, and minimally explored instrumental and applicational strategies. If the current aforementioned bottlenecks are studied and optimized, chiral SFC certainly has a thriving future in enantiomer assessments in various drug-related fields, as highlighted by recently reported applications in the fields of drug development, clinical research, forensic toxicology, and environmental studies.
References
- D. Speybrouck and E. Lipka, J. Chromatogr. A 1467, 33–55 (2016).
- G. Roskam, B. van de Velde, A. Gargano, and I. Kohler, LCGC Europe 35(3), 83–92 (2022).
- C. West, TrAC Trends Anal. Chem. 120, 115648 (2019).
- M. Gumustas, S.A. Ozkan, and B. Chankvetadze, Curr. Med. Chem. 25(33), 4152–4188 (2018).
- L.C. Harps, J.F. Joseph, and M.K. Parr, J. Pharm. Biomed. Anal. 162, 47–59 (2019).
- C.L. Barhate et al., Chem. Commun. 53(3), 509–512 (2017).
- A.S. Breitbach et al., J. Chromatogr. A 1427, 45–54 (2016).
- D. Roy and D.W. Armstrong, J. Chromatogr. A 1605, 360339 (2019).
- L. Nováková and M. Douša, Anal. Chim. Acta 950, 199–210 (2017).
- K. Zawatzky, M. Biba, E.L. Regalado, and C.J. Welch, J. Chromatogr. A 1429, 374–379 (2016).
- L. Zhang et al., J. Supercrit. Fluids 107, 129–136 (2016).
- P.A. Pandya, P.A. Shah, and P.S. Shrivastav, J. Chromatogr. A 1625, 461299 (2020).
- R. Wang et al., RSC Adv. 4(90), 49257–49263 (2014).
- L. Nie, Z. Dai, and S. Ma, J. Anal. Methods Chem. 2016, 1–5 (2016).
- M. Goel, E. Larson, C.J. Venkatramani, and M.A. Al-Sayah, J. Chromatogr. B 1084, 89–95 (2018).
- L. Chen, B. Dean, H. La, Y. Chen, and X. Liang, Int. J. Mass Spectrom. 444, 116172 (2019).
- H. Licea-Perez and C.A. Evans, Anal. Methods 9(17), 2603–2610 (2017).
- C. Su et al., J. Sep. Sci. 40(4), 1010–1016 (2017).
- H. Licea Perez, D. Knecht, and C.A. Evans, J. Chromatogr. B 1009–1010, 7–16 (2016).
- R. Hofstetter, G.M. Fassauer, and A. Link, J. Chromatogr. B 1076, 77–83 (2018).
- G.M. Fassauer et al., J. Pharm. Biomed. Anal. 146, 410–419 (2017).
- M.K. Parr et al., Food Addit. Contam. Part A 34(4), 525–535 (2017).
- D. Folprechtová et al., J. Chromatogr. A 1637, 461846 (2021).
- M. Losacker et al., J. Anal. Toxicol. 45(9), 985–992 (2021). doi:10.1093/jat/bkaa156
- Opiumwet: https://wetten.overheid.nl/BWBR0001941/2020-01-01#BijlageI>
- L. Li, Forensic Chem. 2, 82–85 (2016).
- S. Hegstad, H. Havnen, A. Helland, O. Spigset, and J. Frost, J. Chromatogr. B 1077–1078, 7–12 (2018).
- H. Havnen, M. Hansen, O. Spigset, and S. Hegstad, Drug Test. Anal. 12(9), 1344–1353 (2020).
- H. Segawa et al., Forensic Sci. Int. 273, 39–44 (2017).
- H. Segawa et al., Forensic Toxicol. 37(1), 145–153 (2019).
- H. Segawa et al., Forensic Sci. Int. 318, 110591 (2021).
- J. Rice, A. Lubben, and B. Kasprzyk-Hordern, Anal. Bioanal. Chem. 412(23), 5563–5581 (2020).
- B. Kasprzyk-Hordern and D.R. Baker, Environ. Sci. Technol. 46(3), 1681–1691 (2012).
- A. Svan et al., J. Chromatogr. A 1409, 251–258 (2015).
- D. Camacho-Muñoz, B. Kasprzyk-Hordern, and K.V. Thomas, Anal. Chim. Acta 934, 239–251 (2016).
- K. De Klerck, C. Tistaert, D. Mangelings, and Y. Vander Heyden, J. Supercrit. Fluids 80, 50–59 (2013).
- K. De Klerck, Y. Vander Heyden, and D. Mangelings, J. Chromatogr. A 1363, 311–322 (2014).
- T.A. Berger, B.K. Berger, and K. Fogleman, Compr. Chirality 8, 354–392 (2012).
- C.J. Welch et al., Chirality 19(3), 184–189 (2007).
- R.S. Hegade and F. Lynen, J. Chromatogr. A 1586, 116–127 (2019).
- J. Lin, C. Tsang, R. Lieu, and K. Zhang, J. Chromatogr. A 1624, 461244 (2020).
- R. Geryk, K. Kalíková, M.G. Schmid, and E. Tesařová, Anal. Chim. Acta 932, 98–105 (2016).
- D. Rossi et al., J. Pharm. Biomed. Anal. 118, 363–369 (2016).
- J. Storch, K. Kalíková, E. Tesařová, V. Maier, and J. Vacek, J. Chromatogr. A 1476, 130–134 (2016).
- D.-R. Wu et al., J. Pharm. Biomed. Anal. 131, 54–63 (2016).
- Y. Zehani et al., J. Chromatogr. A 1467, 473–481 (2016).
- X. Bu, E.L. Regalado, J. Cuff, W. Schafer, and X. Gong, J. Supercrit. Fluids 116, 20–25 (2016).
- C.J. Venkatramani et al., Talanta 148, 548–555 (2016).
- Y. Zehani, L. Lemaire, R. Millet, and E. Lipka, J. Chromatogr. A 1505, 106–113 (2017).
- C. West et al., J. Chromatogr. A 1499, 174–182 (2017).
- E. Forss et al., J. Chromatogr. A 1499, 165–173 (2017).
- T. Rogez-Florent et al., J. Pharm. Biomed. Anal. 137, 113–122 (2017).
- M. Iguiniz, E. Corbel, N. Roques, and S. Heinisch, J. Pharm. Biomed. Anal. 159, 237–244 (2018).
- D. Folprechtová et al., J. Chromatogr. A 1612, 460687 (2020).
- R.K. Hofstetter et al., Molecules 24(10), 1927 (2019).
- D. Roy, M.F. Wahab, M. Talebi, and D.W. Armstrong, Green Chem. 22(4), 1249–1257 (2020).
- L.J. Wilson, C. Mi, and C.M. Kraml, J. Chromatogr. A 1634, 461661 (2020).
- S. Declerck, Y. Vander Heyden, and D. Mangelings, J. Chromatogr. A 1624, 461201 (2020).
- P.A. Pandya, P.A. Shah, and P.S. Shrivastav, J. Pharm. Anal. 11(6), 746–756 (2020). doi:10.1016/j.jpha.2020.12.005
- A. Bajtai et al., J. Pharm. Biomed. Anal. 193, 113724 (2021).
- T. Choppari et al., Chromatographia 84(3), 297–306 (2021).
- J. Crépier, E. Corbel, J.-M. Lerestif, A. Berthod, and S. Heinisch, J. Pharm. Biomed. Anal. 202, 114142 (2021).
- Y. Gou et al., Biomed. Chromatogr. 35(12), 1–9 (2021). doi:10.1002/bmc.5211
- Z. Yang et al., J. Chromatogr. B 1020, 36–42 (2016).
- H.Y. Eom et al., J. Pharm. Biomed. Anal. 117, 380–389 (2016).
- S. Hegstad, H. Havnen, A. Helland, B.M. H. Falch, and O. Spigset, J. Chromatogr. B 1061–1062, 103–109 (2017).
- L. Chen, B. Dean, and X. Liang, Bioanalysis 11(4), 251–266 (2019).
- D. Albals, Y. Vander Heyden, M.G. Schmid, B. Chankvetadze, and D. Mangelings, J. Pharm. Biomed. Anal. 121, 232–243 (2016).
- N. Kolderová, et al., J. Chromatogr. A 1625, 461286 (2020).
Gerry Roskam, Bas van de Velde, Andrea Gargano, and Isabelle Kohler are with the Centre for Analytical Sciences Amsterdam, in Amsterdam, in the Netherlands. Roskam and Gargano are also with the van ‘t Hoff Institute for Molecular Science, at the University of Amsterdam. Roskam, van de Velde, and Kohler are also with the Division of BioAnalytical Chemistry in the Amsterdam Institute of Molecular and Life Sciences, at Vrije Universiteit Amsterdam. Direct correspondence to: i.kohler@vu.nl or a.gargano@uva.nl





 Methods for Environmental Analysis")

; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")




. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

