Strategies for The Purification of Synthetic Products in The Pharmaceutical Industry
LCGC Europe
By taking advantage of the benefits provided by normal-phase mode, highly productive and cost-effective strategies for high-throughput purification of drug discovery products have been developed in the analytical laboratories at Lilly-Spain. The straightforward scaling-up of generic protocols from an analytical to a preparative scale has yielded successful results not only when working in HPLC but also when transferring conditions to other standard low and medium pressure chromatographic systems that are routinely used by synthetic chemists.
Synthetic chemists are continuously driven to deliver larger numbers of compounds for screening in the competitive race to reduce the cost and time taken for new bioactive compounds to reach the market. In an effort to meet the quality required for the target products, there is a demand on optimizing purification processes as they have become a main bottleneck and are sometimes more challenging and time-consuming than the setting up of the chemical reaction. Normal-phase chromatography emerges as the elution mode of choice as it offers advantages such as better selectivity for many isomers, minimal drying down or better solubility and stability compared with aqueous solutions.



This work focuses on highly productive and cost-effective strategies developed for normal-phase high-throughput purification of drug discovery products. The straightforward scaling-up of procedures from an analytical scale to gram amounts has been demonstrated not only with standards but also with real-life samples. Instrumentation such as HPLC and other pressure chromatographic systems has been implemented and generic protocols have been developed for the different needs of synthetic chemists in medicinal and combinatorial environments.
Introduction
One of the main goals of pharmaceutical companies is developing new products in shorter times and with lower costs. As more compounds have to be evaluated, the need for both synthetic and analytical knowledge of the compounds to be prepared grows. On the one hand, chemists are developing more complex targets with an increasing number of functional groups in a single molecule. On the other, the regulatory agencies continue to ask for more stringent purity requirements. As a result, purification has become a crucial step in the process of drug development and analytical departments are permanently involved in addressing those challenges.1
Traditionally, organic chemists have been using techniques such as crystallization, liquid-liquid extraction or flash chromatography. The latter has become one of the most commonly used techniques. However, the procedure of choosing the appropriate stationary and mobile phases as well as tracking the compounds in the collected fractions is time-consuming. Furthermore, the bench scale-up relies mostly on "trial and error" processes and on few "rules-of-thumb". As a result, the isolation of the desired product from the crude mixture is often incomplete and several steps must be used to match purity requirements. Unfortunately, each step results in a waste of sample and resources (time and cost).
In an ideal state, a specialized group could centralize all purifications coming from a research laboratory. With current resources, centralization will result in an additional bottleneck adversely affecting the analytical groups so that problems are not faced in time. Although most of the purity assessments are performed under reversed-phase (RP) high performance liquid chromatography–mass spectrometry (HPLC–MS) conditions, normal-phase (NP) chromatography is still the method of choice for isolation purposes, mainly at large scale. Consequently, the Lilly-Spain analytical group has made a great effort to speed up method development time and reduce operating costs of NP analytical and purification processes and also to deploy our protocols to the different synthetic groups.
Experimental
All the analytical studies were performed on a Series 1100 liquid chromatography/mass selective detector LC/MSD (Agilent, Waldbronn, Germany) driven by LC/MSD ChemStation Rev. A.10.02 (1757) © 1990–2003 Agilent Technologies. This system is composed of a solvent degasser (G1379A), a quaternary pump (G1311A), an autosampler (G1313A), a column compartment (G1316A), a diode array detector (G1315B) and a mass spectrometer (G1946D). An evaporative light-scattering detector (Polymer PL-ELS 1000) is coupled in series. Flow from a UV detector is split for MS and ELS. For automation of the system, a Cheminert HPLC 8 column selector (VALCO) was controlled via a remote logic level signal. Analyses were run on 5 μm, 4.6 mm i.d. × 250 mm units. Flow-rate in all instances was 1 mL min-1 . For the gradient elution programme, composition of the mobile phase went from 10 to 40% of the polar component in 15 min in all instances but from 2 to 8% methanol for the dichloromethane (DCM)/methanol mixtures to avoid damage of unmodified silica columns. For UV monitoring, 215, 254, 280, 325 and 375 nm wavelengths are selected by default (diode array detector). In most of the situations except for the DCM mixtures (because of the cut off of the solvent), material quality is defined on the basis of the signal at 215 nm.
Semi-preparative HPLC purifications were performed using a Waters Delta Prep 4000 liquid chromatograph (Waters, USA) with a diode array detector and controlled through the Mass Lynx v3.5 software package. Purifications were conducted with 7 μm, 250 × 20 mm column. Standard flow-rate was 18 mL min-1 .
Preparative HPLC purifications were processed using a Novasep LC-50 system driven by HIPERSEP LAV PLC software version 1.1.4 and PC software version 1.1.6. The two detectors, variable ultraviolet and refractive index, were coupled in series. A 10 μm, 80 × 180 mm dynamic axial compression column was used for this purpose. Standard flow-rate was 200 mL min-1 .
Lower flow-rates were sometimes applied to address purification of extremely complex mixtures (usually under isocratic elution mode).
The names and structures of Lilly compounds used to illustrate chromatographic separations cannot be disclosed because of proprietary reasons.
Results and Discussion
Analytical HPLC/MS/ELSD screen: Traditionally, appropriate selectivity and retention has been determined by experimenting with different solvent combinations and concentrations on thin-layer chromatography (TLC) plates. The main limitation is that, in the high-throughput environment, this tool gets neither quantitative nor qualitative accurate data about the purity of the compounds monitored. In some instances, co-elution of related compounds is not detected and purification fails to achieve the purity required.
Because it is universally accepted that RP-HPLC–MS is the tool of choice for purity assessments, the Lilly analytical group addressed the challenge of developing and implementing a similar NP-HPLC–MS screen for both the quality control and method development for purification purposes.2 In contrast to electrospray ionization-mass spectrometry (ESI-MS), the atmospheric pressure chemical ionization (APCI) interface showed good compatibility to organic solvents used in normal phase. Furthermore, evaporative light-scattering detection (ELSD) was also investigated as an alternative for routine analysis.
Automation was given high priority to increase productivity. An eight column-switching device, with five unmodified silica and three modified silica units, was set up. Following data reported in literature,3 selection of standard mobile phases was based on the hydrogen bonding and dipole properties of organic solvents. On these bases, solvents from the different corners of the so-called selectivity triangle were selected for maximum change in selectivity. Hence, mixtures of alkane (n-hexane) with ether (methyl-tert-butyl ether), ketone (acetone) and alcohol (ethanol or isopropanol) were defined by default. In addition, a DCM/alcohol mixture was also selected for addressing those types of compounds that are highly retained and/or show poor solubility under normal-phase chromatographic conditions. Trifluoroacetic acid was selected as a modifier for processing acidic compounds on unmodified silica, and it was added to n-hexane at 0.05% (v/v). For basic compounds, however, evaluation of modified silica-NH2 with mixtures of alkane (n-hexane) and an alcohol (ethanol or isopropanol) was the first choice.
Complete elution of compounds within the gradient time is an important issue for any automated screening to avoid cross-contamination between sequential injections. Although wide gradients and a clean up step at the end of a run are usually applied in reversed-phase chromatography without affecting analysis cycle time, normal-phase analysis requires a different approach to ensure reproducible results and reduce re-equilibration times. To address those challenges, a robust tailored gradient programme was defined. Some authors have recently defined normal-phase gradient elution as a reproducible process with short re-equilibration times, as long as solvent combination is carefully selected.4–6 The Lilly analytical group agrees with this statement. Its optimized gradient programme included studies to determine the best initial and final mobile phase composition and the optimum gradient steepness. This was to first ensure the reproducibility and resolution of the analyses and secondly, to minimize the risk of unexpected results caused by gradual deactivation of the stationary phase and solvent demixing effects. For all mobile phases, except mixtures of DCM/methanol, the standard elution programme was fixed as a 10 to 40% polar component gradient in 15 min. In addition, each unmodified silica column was assigned a different but dedicated solvent system to minimize re-equilibration time.
A few months ago the group published the standard normal-phase chiral HPLC–MS protocol developed and implemented in its analytical laboratories.7 In this work, the successful direct linking of an atmospheric pressure ionization mass spectrometer, MS-APCI, with a high performance liquid chromatography/diode array detector (HPLC/DAD) system was reported. These operated under normal-phase mode and without post-column addition of MS-compatible solvents. This methodology provides the high specificity/selectivity (identification of target compounds in complex mixtures) and accuracy (1–2% area level) required for daily studies and has proven its robustness for more than three years. As a result of this success, identical configuration of the MS detector has been defined in non-chiral normal-phase HPLC methodology that is described in this article.
Results achieved using these standard gradient protocols allow not only the best solvent combination for selectivity (qualitative) to be defined but also the most appropriate ratio (quantitative) for retention adjustment for preparative HPLC. As a result, the number of experiments required to find optimum conditions for the highest resolution is minimized.
From HPLC analysis to HPLC purification: HPLC is often the only choice to fulfil the requirements of product purity demanded in pharmaceutical industries. Whereas analytical HPLC focuses on resolving and identifying each mixture's compound, the economical task of preparative chromatography implies finding the conditions to achieve the required purification degree with minimum cost and maximum yield. Factors such as resolution of all components of the mixture, spectroscopic characteristics of the solvents (UV transparency), volatility (removal from isolated fractions) and solubility properties (maximum sample load) need to be evaluated.
By checking retention achieved in the gradient screen and, therefore, mobile phase composition required for elution, scale-up is direct and does not require optimization steps. Routinely, tailored gradient elution (usually less than a 20% increase of polar component in 10 min depending on analytical resolution, scale of process and sample loading) is selected for achieving shorter elution times, greater sample loading and minimum fraction collection volume. A simple rule to easily define a gradient range that achieves appropriate resolution of the peaks of interest (either two target compounds or a target compound and its nearest impurity) within 10 min gradient time can be applied by non-specialized researchers. Therefore, for two compounds, A and B, with retention times in the standard analytical gradient that correspond to X and Y percentages of the polar component of the mobile phase, respectively, the gradient will go from (X-10) to Y in 10 min. Figure 1 displays two examples of the standard gradient for batch purification.

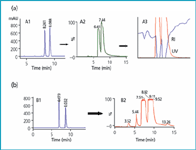
Figure 1: From HPLC analysis to HPLC purification. (a) HPLC isolation of a mixture of the standards acetophenone and dimethylphthalate. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm for A1; 7 μm, 20 à 250 mm for A2 and 10 μm, 80 à 180 mm for A3); hexane/MTBE; gradient from 10 to 40% MTBE in 15 min for A1 and from 15 to 30% MTBE in 10 min for A2 and A3; flow-rate: 1, 18 and 150 mL min-1 for A1, A2 and A3, respectively; UV detection: 215 nm for A1 and 280 nm for A2/A3. Sample loading: 100 mg and 1.8 g (0.9 g each standard) in A2 and A3, respectively. (b) HPLC isolation of a pharmaceutical compound synthesized in our research laboratories. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm for B1 and 7 μm, 20 à 250 mm for B2); hexane/2-propanol (IPA); gradient from 10 to 40% IPA in 15 min for B1 and from 15 to 25% IPA in 15 min for B2; flow-rate: 1 and 18 mL min-1 for B1 and B2, respectively; UV detection: 215 nm. Sample loading in B2 is 200 mg.
Nevertheless, if resolution is poor or the amount of sample to be processed is too large, different strategies are applied. The most common protocols followed in Lilly's laboratories are summarized as follows:
Stacked injections: This approach helps save time and cost because sequential injections are done before complete elution takes places. Figure 2's example compares the results afforded by standard batch chromatography and stacked injections during the purification of a compound synthesized in its research laboratories. While the former requires 1 h completing the batch, the latter achieves the same goal in less than 30 min.


Figure 2: Stacked injections. Purification of a pharmaceutical compound synthesized in our research laboratories. Conditions: Kromasil Si 60 (7 μm, 250 à 20 mm); hexane/acetone 8/2; flow-rate 18 mL min-1 ; UV detection: 235 nm. For a 36 mg batch chromatography total elution of highly retained impurities takes 18 min. (a) The time required for purification of the 110 mg batch would be almost 1 h. Applying stacked injections (b), a new 36 mg fraction of sample is injected each 6 min and the time needed to complete the purification is reduced to 30 min. Presence of impurities precluded larger sample loading.
Recycling technology and peak shaving: The effectiveness of any purification dramatically decreases when a difficult separation has to be accomplished and/or the resolving capacity of the chromatographic system is poor. The recycling technique offers a solution for those challenges. As the sample elutes from the outlet of the column, the sample is re-directed to the inlet of the column, therefore, the column length is artificially increased, the resolution is highly improved and the solvent consumption is minimized. Because the application of this technology requires the use of isocratic elution, a certain grade of peak dispersion can sometimes be observed because of known factors such as the nature and dilution of the solute, properties of the stationary phase or flow-rate. Nevertheless, in all instances, there is a substantial net gain in resolution. Furthermore, combination of recycling and peak shaving, if possible, dramatically increases throughput. As a result of these benefits, easy-to-use methodologies are applied daily in its laboratories. See Figure 3 for representative examples.

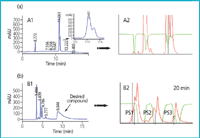
Figure 3: Recycling technology and peak shaving. (a) HPLC purification of an acidic intermediate prepared in our research laboratories using recycling methodology to increase resolution. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm for A1 and 10 μm, 80 à 180 mm for A2); Hexane-TFA 0.05%/2-propanol (IPA); gradient from 10â40% IPA in 15 min for A1 and isocratic 25% IPA for A2; flow-rate 1 mL min-1 for A1 and 135 mL min-1 for A2; UV detection: 215 nm. Sample loading for A2: 0.7g. Productivity: 2.1g/h. (b) HPLC purification of a synthetic intermediate prepared in our research laboratories using peak shaving and recycling methodology to increase productivity. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm for Figure 3(a) and 10 μm, 80 à 180 mm for B1); hexane/2-propanol (IPA); gradient from 10 to 40% IPA in 15 min for B1 and isocratic 10% IPA for B2; flow-rate 1 mL min-1 for B1 and 135 mL min-1 for B2; UV detection: 215 nm for B1 and 235 nm for B2. Peak shavings: PS1, 2 and 3. Sample loading: 7 g. Productivity: 21 g/h.
Choice of detection: When a chromatographic method is developed for purification purposes, the balance between the need for a solvent system that provides adequate resolution of all components in the mixture and the need for detector parameters that provide good sensitivity and low solvent background absorbance is carefully evaluated. A complex purification can be more easily addressed by simply selecting the specific UV wavelength signal to monitor the process. Selection of a standard wavelength as 215 or 254 nm can preclude selective monitoring of elution, therefore collection, and also saturate UV detection even under non-overloading conditions. In contrast to analytical studies, sensitive detectors are not required in preparative LC because solute concentrations are high. Traditionally, 215 nm is the preferred signal for standard purity assessment of complex mixtures for all compounds (except those lacking chromophores), because of the excellent UV response at this wavelength. However, selection of such a low wavelength in prep scale, where a high concentration of sample is injected, causes problems because it is difficult to determine whether the overlapping peaks seen are because of an overloaded column or a non linear detector response. Careful definition of the most selective wavelength is therefore performed before starting the purification process. The wavelength selected is not always the maximum absorbance of the target compound, but the most selective wavelength to discriminate between the target compound and the main close impurities. In our experience, the selection of an adequate method of detection is essential and can make a difference between the success and failure of the processes. Figure 4 shows results achieved with a mixture of the standards warfarin and nifedipine. Even after applying a very fast gradient and column overloading, the two compounds can easily be isolated with high recovery and purity by selecting 325 and 375 nm as UV monitoring signals. UV detection is the most common tool because of its versatility, sensibility, easy handling and good results. It fails for those compounds lacking chromophores and also if large amounts of material need to be processed (the column is overloaded and, therefore, the UV detector is saturated). For those situations, a refractive index (RI) detector is coupled on-line. In the example represented in Figure 5, monitoring of the process by RI allowed 5-fold sample loading compared with UV detection.

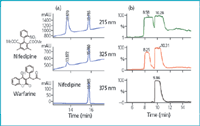
Figure 4: Choice of detection and selection of an appropriate UV wavelength. Semi-prep HPLC purification of a mixture of warfarin and nifedipine. Selection of UV wavelength allows increasing sample loading without sacrificing purity. Conditions: Kromasil Si 60 (5 mm, 4.6 à 250 mm for (a) and 7 μm, 20 à 250 mm for (b); hexane/acetone; gradient from 10 to 40% acetone in 15 min for (a) and for 25â40% acetone in 10 min for (b); flow-rate 1 mL min-1 for (a) and 18 mL min-1 for (b). Sample loading: 200 mg.
From analytical HPLC to radial compression systems and low-pressure chromatography: Unfortunately, the availability of a large number of HPLC systems in synthetic laboratories is not very common. Nevertheless, organic chemists are asked to complete more projects in less time with limited resources. Consequently, producing pure products in a timely manner requires more effective purification strategies. In recent years, manufacturers have got involved in this challenge by providing new instrumentation and techniques with remarkable capabilities (medium radial compression and low-pressure chromatography systems) that have positive implications in terms of cost, versatility and sample throughput. With all these techniques available, analytical studies become the key step as a high quality method development will guarantee a successful result. With the fundamental theories and methods derived from the HPLC screen, standard protocols have been defined to transfer conditions successfully from its high performance systems to any of the medium-low pressure chromatographic systems available in its synthetic research laboratories. The properties of the stationary phase (metal content, particle size, packing density etc...) are important in chromatography. However, the specific devices for sample loading or maintaining a bed's stability (such as radial compression) also play a crucial role in the chromatographic process. For these reasons, parameters such as selectivity, resolution and retention of the different components of the mixture in analytical HPLC experiments have been evaluated to define the most effective purification methodology and instrumentation that will achieve the highest yield and minimum cost. Following its protocols, complete analytical HPLC method development for successful extrapolation to purification processes in other pressure chromatography systems can be developed in as little as 1 h. To develop a robust and easily transferable methodology the group focused on defining the optimum analytical HPLC retention and resolution for the desired compound and its nearest impurities. This ensured a successful transfer even though it meant working with different silica properties and operating pressures. Studies showed that the appropriate retention factor, k', in a 4–6 range (t0 3.5min) could be defined. Resolution required for separation will be highly dependent on the structural properties of the molecule (easily identified by peak width and symmetry in analytical HPLC studies) as well as the sample loading desired for purification. In general, Δk' 1 between the target compound and its closest impurities will afford successful results.

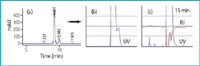
Figure 5: Choice of detection and RI detector guide. Preparative HPLC purification of a synthetic intermediate prepared in our laboratories. Monitoring of the process by RI allowed 5-fold sample loading compared to UV detection. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm for (a) and 10 μm, 80 à 180 mm for(b/c); hexane/ethanol 98/2; flow-rate 1 mL min-1 for (a) and 135 mL min-1 for (b/c). Sample loading: 0.5 g for (b) and 2.5 g for ©.
Radial compression chromatography: Prepacked cartridges and radial compression technology create stable, effective separation beds that give a high degree of confidence in the consistency and scalability of preparative chromatography processes. For this reason, this chromatography is the first choice for scale-up of separation solutions for complex purification problems. Figure 6 illustrates the development of conditions for large-scale medium radial compression chromatography purification of a synthetic compound prepared in its laboratories. Selection of the appropriate solvent combination by an HPLC screen was followed by adjusting the mobile phase composition to match the required chromatographic parameters. Identification of the components in the mixture is achieved by UV/MS and reproduced by TLC ensuring that a single spot corresponds to a single compound, therefore, allowing both accurate monitoring and quality control of the target product.


Figure 6: Radial compression chromatography. HPLC method development for medium radial compression chromatography purification of a synthetic intermediate prepared in our laboratories. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm); hexane/MTBE; gradient from 10â40% MTBE in 15 min for (a), isocratic 30% MTBE for (b) and isocratic 5% MTBE for (c). While conditions shown in (b) are optimal for HPLC purification purposes, they will fail to achieve resolution in other pressurized systems. To define appropriate resolution for low- and medium-pressure chromatography, we need to "sacrifice" speed/theoretical plates in HPLC chromatography (c). Applying conditions so defined (c), a 90 g batch was purified in medium radial compression chromatography 75 M in a single experiment.
Automated low-pressure chromatographic systems: The selection of the suitable separation technique and media strongly influences productivity and economy. Low-pressure systems offer many of the benefits of automated flash chromatography in a fast and easy-to-use system for milligram- to gram-scale purification of samples. Among them, the parallel system brings a breakthrough in high-throughput parallel purification as it offers ten channels with individual gradient control on each one, thereby dramatically improving the productivity over traditional methods. This strategy can also be successfully applied in those instruments. Figure 7 shows the parallel purification of ten mixtures in less than 5 min. RP-HPLC–MS quality control of all compounds isolated in this way confirmed purity of greater than 97%.


Figure 7: Automated low-pressure chromatography. HPLC method development for parallel purification of acidic intermediates prepared in our laboratories. Conditions: Kromasil Si 60 (5 μm, 4.6 à 250 mm); hexane-TFA 0.05%/acetone; gradient from 10â40% acetone in 15 min at 1 mL min-1 for (a). The lack of knowledge on the pka range for those compounds and the need of adding trifluoroacetic acid as modifier prompted us to run a preliminary experiment at small scale in our analytical laboratories to confirm the generality of our methodology (b): RediSep 4 g, 25% acetone, 18 mL min-1 ). The method was successfully applied in parallel by the chemist to ten different mixtures (c): RediSep 12 g, 25% acetone, 30 mL min-1 , 300â500 mg/batch).
SPE: Prefractionation of complex samples is increasingly recognized as essential for accelerating drug discovery processes. For this purpose, few techniques are as easy, inexpensive and quick as solid-phase extraction (SPE). SPE offers improved selectivity and specificity, efficient extraction, quantitative recoveries and very low solvent consumption. Taking advantage of all these properties, it can not only be used for pretreatment of samples (to avoid blockage of HPLC because of undesired retention of highly polar impurities) but also to isolate target compounds with high purity requirements. As shown in previous examples, this protocol can also be successfully transferred to SPE. However, special care needs to be taken concerning control of the flow-rate to ensure reproducible results. The remaining chromatographic requirements concerning cartridge selection and conditioning/elution conditions (e.g., column volume) just follow manufacturer's recommendations. The use of SPE in its laboratories is illustrated in Figure 8. Quick SPE filtration of raw material increased the purity level from 19–75% and decreased sample amount from 1.1 g to about 300 mg, therefore, saving time and solvent and facilitating applicability of stacked injections for further HPLC treatment.

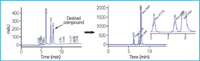
Figure 8: SPE purification. SPE prepurification of an intermediate prepared in our laboratories. Conditions: Luna NH2 (5 μm, 4.6 à 250 mm); hexane/ethanol; gradient from 10â40% ethanol in 15 min at 1 mL min-1 . Figure 1(a) displays analysis of the raw material (1.1g, 19% purity) while (b) corresponds to quality control of the target compound after SPE treatment (300 mg, 75% purity). SPE conditions: Strata NH2 (10 g), column volume (CV) was 30 mL, elution with hexane (1CV) was followed by elution with hexane/EtOH 98/2 (1CV), hexane/EtOH 95/5 (1CV) and hexane/EtOH 9/1 (CV). Desired compound was mainly collected in the 9/1 fraction.
Conclusions
Addressing the purification problem with specific strategies and implementing technical solutions and logical protocols in every laboratory will assure the success of production process performance and reduce the development time and economical cost to market new products.
Different separation techniques based on normal-phase elution have been optimized in Lilly-Spain to speed up the purification process. Chromatographic parameters (retention, resolution or selectivity), sample requirements (number, amount, solubility or final purity) and operating conditions (stacked injections, recycling, selection of wavelength signal and operating pressure) are carefully evaluated in its daily work to achieve successful results. A smooth scale-up procedure based on a well-designed transfer assures product quality, cost effectiveness and timely achievement of drug discovery objectives. Finally, but no less important, introducing, educating and updating chemists in the field of chromatography has been a crucial goal of this work.
References
1. A. Brandt and S. Kueppers, LCGC Eur., 15(3), 147–151 (2002).
2. M.L. de la Puente (unpublished results).
3. J.L. Glajch, J.J. Kirkland and L.R. Snyder, J. of Chromatogr., 282, 269–280, (1982).
4. V. R. Meyer, J. Chromatogr. A, 768, 315–319, (1997).
5. P. Jandera, J. Chromatogr. A, 797, 11–22, (1998); 845, 133–144, (1999) and 965, 239–261, (2002).
6. P. Renold, E. Madero and T. Maetzke, J. Chromatogr. A, 908, 143–148 (2001).
7. M.L. de la Puente, J. Chromatogr. A, 1055, 55–62 (2004).
Pilar López Soto-Yarritu is a research associate chemist in the Analytical Technologies Department at Lilly Alcobendas DCRT. She is focused on the achiral normal phase chromatography area. Amelia González is a research associate chemist in the Analytical Technologies Department at Lilly Alcobendas DCRT. She is focused on the chiral normal phase chromatography area. Leticia Cano is a technician in the Analytical Technologies Department at Lilly Alcobendas DCRT. She is focused on the achiral normal phase chromatography area. María Luz de la Puente, PhD is a senior research scientist in the Analytical Technologies Department at Lilly Alcobendas DCRT. She currently leads a group of 5 scientists focused on normal phase chiral and non-chiral areas.













Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

