Simultaneous Determination of Synthetic Pharmaceutical Peptides and the Acetate Counterion by Mixed-Mode Weak-Anion-Exchange Chromatography
LCGC North America
Oxytocin acetate samples were analysed successfully for both the peptide and acetate with a single method using a mixed-mode weak-anion-exchange column.
Peptides perform a wide variety of physiological functions and are critical to regulatory and cell signaling pathways. As such, many peptides have been investigated as therapeutic drugs and are active vasodilators, vasoconstrictors, hormones, and neuropeptides. Any peptide under consideration as a therapeutic drug must be analyzed to determine both the peptide content and any counterions that might be present.
Oxytocin is a neuropeptide with therapeutic uses including stimulating labor, control of postpartum hemorrhage, and inducing lactation (1). This nonapeptide has a disulfide bond between two cysteine residues forming a large ringed structure (Figure 1) that is stable between pH 3 and 5. In more acidic solutions, the peptide hydrolyzes, whereas in neutral and basic conditions it produces dimers and polymeric mixtures by the creation of intermolecular disulfide bridges that lead to peptide deactivation (2).


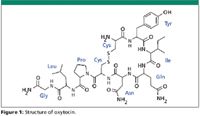
Figure 1
As a commercial product, oxytocin is available as an acetate salt. The USP and EP allow 6–10% by weight of acetic acid in the solid peptide drug. The presence of acetic acid as a counterion in peptide therapeutics is common and is found in numerous small peptides. Current USP and EP monographs use two separate methods to assay oxytocin and the acetate counterion (3–6). The USP monograph to assay oxytocin uses a gradient of sodium phosphate and acetonitrile on an L1 (C18) column with a 20 min run time and a flow rate of 1.5 mL/min. The EP oxytocin assay monograph is similar to the USP monograph, in terms of the column type and mobile phase. However, the EP method specifies a 1.0-mL/min flow rate and a 30-min run time with an additional 15 min of equilibration. The current USP and EP chapters for acetic acid in peptides are equivalent and are not specific to acetic acid in oxytocin. Like the oxytocin assay method, an L1 column is used, in this case with a phosphate buffer–methanol gradient. This acetic acid method has a run time of 22 min at a flow rate of 1.2 mL/min and uses a steep gradient to wash the peptide from the column after the acetate is eluted. To fully analyze oxytocin acetate by the USP methods takes a minimum combined total of 42 min and two separate injections with different methods.
Acetic acid is retained weakly on an L1 column and quantification can be challenging, particularly if other compounds that elute near the void volume are present. Because of this weak retention of acetic acid, the rapid simultaneous determination of oxytocin and acetate in a single injection on an L1 column is difficult. Chromatographic conditions that retain both acetic acid and resolve the peptide and related impurities lead to long run times compared to current high performance liquid chromatography (HPLC) methods. Alternative methods to assay neuropeptides such as enzymatic assays, spectroscopic methods, gas chromatography, capillary electrophoresis, mass spectrometry, and ion chromatography effectively measure either the peptide or the acetic acid, but have inherent difficulties in simultaneously determining the peptide and an acetate counterion (7–9).
Conversely, HPLC separation on a mixed-mode weak-anion-exchange column retains acetate, allowing better quantification of the low UV absorbing acetate ion and also retains the peptide of interest. By using pH to control the selectivity of the column, a single method can be developed for simultaneously determining peptides and acetate counterions (10).
Materials and Methods
Materials: Oxytocin, angiotensin I, bradykinin, neurotensin, and tetra-sodium pyrophosphate decahydrate were purchased from Sigma-Aldrich (St. Louis, Missouri). Potassium phosphate, monobasic was purchased from Fisher Scientific (Hampton, New Hampshire). Acetonitrile, chlorobutanol, methanol, phosphoric acid, and pH buffers were purchased from VWR Scientific Products (West Chester, Pennsylvania). Glacial acetic acid was purchased from USP (Rockville, Maryland). Deionized water, type 1 reagent grade, 18 MΩ-cm resistivity or better was used throughout.
Reversed-phase HPLC: Aliquots (25 μL each) of sample were injected onto an UltiMate 3000 HPLC system (Dionex Corp., Sunnyvale, California) with a mixed-mode weak-anion-exchange reversed-phase column (150 mm × 2.1 mm, 5-μm Acclaim Mixed Mode WAX-1 (Dionex Corp.) at a flow rate of 210 μL/min. The peptides were eluted with a 36-min gradient as follows: 98% 50 mM potassium phosphate at pH 4.2 (A), 2% methanol (B) for 5 min, 2–50% B in 20 min, 50% B for 10 min, 5 min of equilibration consisting of 2% B before injection. The column temperature was maintained at 30 °C. Detection of eluted peaks was accomplished by a variable-wavelength UV–vis detector set to 220 nm.
The USP monographs for oxytocin and acetic acid in peptides were repeated using a 150 mm × 2.1 mm, 5-μm Acclaim 120 C18 column (Dionex Corp.).
Sample and standard preparation: Acetic acid standards: A 20.0-mg/mL stock solution of acetic acid was prepared and stored at 4 °C. Working standards were prepared daily from the stock solution.
Peptide samples: Peptide stocks were prepared by dissolving 1.0 mg of peptide in 1.00 g of deionized water. These stocks were further diluted to 250 μg/mL with mobile phase A before analysis. Stocks and samples were stored at 4 °C.
Oxytocin with chlorobutanol: 250 μL of a 1.0-mg/mL stock solution of oxytocin was diluted to a total volume of 1 mL in 95:5 50 mM potassium phosphate (pH 6.0)–methanol containing 0.5% chlorobutanol.
Results and Discussion
Separation: Figure 2 shows an example of the separation of acetic acid and oxytocin using the USP oxytocin assay (3). Acetic acid is retained weakly on the column under the conditions used for the oxytocin assay. Quantification of acetic acid at concentrations present in the peptides would be very difficult, particularly if there are other poorly retained components in the drug formulation. The chapter method to determine acetic acid in peptides has better retention of acetate, but can be prone to interference, as shown in Figure 3. The acetic acid present in the peptide is difficult to determine due to a closely eluting peak from an impurity in oxytocin. Spiking the sample with acetic acid confirms the presence of acetic acid and further illustrates the difficulty in quantifying acetate in peptide samples. The acetic acid method cannot be used to quantify oxytocin due to low resolution between the peptide and neighboring peaks.


Figure 2
In comparison, Figure 4 shows the separation of acetic acid and oxytocin in a peptide sample on a mixed-mode column. The pH of the mobile phase was optimized at 4.2 after investigating a range of 3–6. A pH of 4.2 offers the best compromise between resolution of peptide impurities, acetate retention time, and total run time. At pH values below 3.5, acetate is retained weakly on the column and pH values of 5–6 caused poor resolution between oxytocin and peptide impurities. At the optimized pH of 4.2, the acetate and oxytocin peaks are resolved from other impurities and easily quantified. The resolution between oxytocin and the next nearest peak is 2.6, greater than the USP monograph requirement of 1.5. A peptide impurity, most likely small amounts of the oxytocin dimer (peak 3), is present in this peptide sample that is eluted at 27 min. If this impurity is absent, the run time can be reduced to 20 min.


Figure 3
Chlorobutanol is a common preservative that is approved for use in injectable oxytocin solutions. To confirm that the presence of this preservative does not interfere with oxytocin or acetate quantification, an oxytocin sample was prepared with chlorobutanol. There is significant resolution between oxytocin and chlorobutanol (USP Rs = 18.16). The USP resolution between oxytocin and the next nearest peak is equivalent to the resolution in the unpreserved sample. The chlorobutanol, with a retention time of 25.23 min does not interfere with determination of acetate or oxytocin.


Figure 4
Quantification Assay Linearity, Limits of Quantitation, and Limits of Detection
Oxytocin: Determination of oxytocin by the EP method currently requires 250 μg/mL of oxytocin (4). The USP monograph assay specifies 10 USP units/mL, which is equivalent to 25 μg/mL of oxytocin assuming an activity of not less than 400 units/mg (3). The linear response of oxytocin determination by detection at 220 nm was investigated from 15 μg/mL to 250 μg/mL to cover the range of both monographs. The correlation coefficient across this range was determined to be 0.9997.
The LOD and LOQ for oxytocin determination were measured by injecting samples that produced a response three and 10 times the measured peak-to-peak noise, respectively. These injections confirmed the estimated LOD of 0.06 μg/mL and estimated LOQ of 0.2 μg/mL for oxytocin. These values are well below the concentrations specified by the EP and USP, ensuring straightforward quantification.


Table I: LOD, LOQ, and linearity for the determination of acetate and oxytocin
Acetate: The concentration of acetate counterion in peptide preparations can vary greatly between peptides. The linearity of the acetate response was tested between 5.0 and 35 μg/mL to cover the expected range of acetate in the peptides investigated. The correlation coefficient, LOD, and LOQ are presented in Table I.


Table II: Determination of acetate in four different peptides
Precision and Accuracy of acetate analysis in representative peptides: The amount of acetate present in the oxytocin sample used for this work is small compared to the amount allowed in the drug product. Containing ~0.5 moles of acetate per mole of oxytocin, the weight percent of acetate is only 2.5%. However, the amount of acetate in peptides can be as high as 10% (w/w). To determine the concentration of acetate in peptide solutions, four peptides were prepared at 250 μg/mL: angiotensin 1, bradykinin, neurotensin, and oxytocin. These peptide samples were each analyzed to determine acetate. Table II presents the peptide precision and acetate determination data for the four representative peptides that incorporate acetate counterions. In all cases, the peptide peak area precisions (RSD) are <0.7, which exceeds the USP criteria for oxytocin. The acetate peak area precisions (RSD) are all <2, exceeding the USP and EP criteria of <5.


Figure 5
Figure 5 illustrates the separation of acetate from the peptides for oxytocin, bradykinin, angiotensin I, and neurotensin. In each case, the acetate and the peptide are quantifiable with good resolution of peptides and impurities using a single injection. Figure 6 shows an expanded view around the acetate peak. The acetate peak is integrated easily with no interferences from other components in the peptide samples.

Figure 6
Recovery of acetate in the four peptides was tested by spiking acetic acid into the peptide solutions, except neurotensin, which was spiked with sodium acetate. The recoveries ranged from 98% to 105% (Table III). These data suggest that the determination of acetate in peptide samples is accurate. Comparison of the lot analysis of acetate in the peptides to that determined is also shown. The determined acetate amounts in the peptide samples are in agreement with the labeled acetate to peptide mole ratio. The concentrations are within ±10% of the label claims or the lot analysis. The neurotensin sample contains 8% acetate, as measured by ion chromatography, based upon the certificate of analysis for the lot. The determined amount of acetate in neurotensin was consistent with the value of 8% acetate, as listed in Table III. Both recovery data and determined acetate amounts in comparison to the suppliers stated amounts show the method to be accurate.


Table III: Recovery of acetate in peptide preparations
Conclusion
The mixed-mode column has been used to determine peptides and the acetate counterion in a single injection simultaneously. The method is fast, accurate, and has the sensitivity needed to determine acetate concentrations in 250 μg/mL peptide samples. The resolution and precision for replicate injections exceed the USP and EP monograph requirements for both oxytocin and acetate. Acetic acid has been quantified successfully in four different peptides, each with a different native acetate concentration, showing the flexibility and broad applicability of this method.
Deanna C. Hurum, Brian M. De Borba, and Jeffrey S. Rohrer
Dionex Corporation, Sunnyvale, California.
Direct correspondence to: deanna.hurum@dionex.com.
References
(1) C.M. Karbiwnyk, K.C. Faul, S.B. Turnipseed, W.C. Andersen, and K.E. Miller, J. Pharm. Biomed. Anal. 48, 672–677 (2008).
(2) F.A. Chaibva and R.B. Walker, J. Pharm. Biomed. Anal. 43, 179–185 (2007).
(3) Oxytocin, The United States Pharmacopeia 31, American Pharmaceutical Association, Washington, DC (2008).
(4) Oxytocin, EP 5.0 01/2005:0780 The European Pharmacopeia, 5th ed., Council of Europe (2005).
(5) Acetic Acid in Peptides <503>, USP 32-NF27, American Pharmaceutical Association, Washington, DC (2010).
(6) Acetic Acid in Synthetic Peptides, EP 2.5.34 The European Pharmacopeia, 5th ed., Council of Europe (2005).
(7) M. Perry, Q. Li, and R.T. Kennedy, Anal. Chim. Acta 653, 1–22 (2009).
(8) M.-J. Rocheleau, Current Pharm. Anal. 4, 25–32 (2008).
(9) I.N. Glazkov, N.L. Bochkareva, I.A. Revel'skii, Russian J. Anal. Chem. 60, 124–136 (2005).
(10) Dionex Corporation. Simultaneous Determination of Pharmaceutical Peptides and Acetate by HPLC with UV detection using the Acclaim Mixed Mode WAX-1 column, Application Note 234, LPN 2291. Sunnyvale, California (2009).













Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.

Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

