The Simple Use of Statistical Overlap Theory in Chromatography
Special Issues
This article explains how statistical overlap theory can be applied to chromatography in everyday usage.
The statistical overlap theory (SOT) of chromatography relates the number of peaks that appear in a chromatogram to the number of detectable components and the peak capacity. This theory transformed chromatography in how it revealed that on a statistical basis the number of peaks underestimates the number of components present in the chromatogram. In this paper, we show how this theory can be applied to chromatography in everyday usage.
The statistical overlap theory (SOT) is a useful theory that gives the relationship between the number of peaks observed in a chromatogram, p, and the number of detectable components, m. This theory, originally devised by Davis and Giddings (1), assumed that these components were distributed using Poisson statistics leading to peaks being distributed randomly across a chromatogram. The results were rather sobering as one of the many predictions of this theory states that “a random chromatogram will never contain more than about 37% of its potential peaks and, worst of all from an analytical point of view, 18% of its potential single-component peaks” (1). In other words, only 18% of the peaks are from single, pure components and only 37% or approximately one-third of the components, show up as unique peaks because of peak overlap. It is also stated “that a chromatogram must be approximately 95% vacant in order to provide a 90% probability that a given component of interest will appear as an isolated peak”.
Simple Derivation
SOT shows that one of the most important parameters in any chromatographic separation is the peak saturation, because it dictates how crowded the separation is. The common label for saturation is α, but it is not the same as selectivity, which often has the same label. For complex biological samples α > 1 and for typical samples with a few components α < 1. For a separation of moderate complexity, α ≈ 1. The key to deciding whether to proceed with a multidimensional separation as opposed to a single column separation is to understand the origin and magnitude of α.
The treatment that follows is based on time as the independent variable. Space (as is the case for thin-layer chromatography, for example) and time are equivalent in this treatment (1). The peak saturation α is a metric of peak crowding equal to


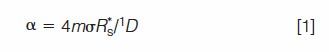
where m is the number of detectable single component peaks (SCPs) with temporal standard deviation σ that occupy a separation space of extent 1D. The term 1D is the time difference between the first and last peaks in a chromatogram. A single component peak is specifically a peak in which a pure component resides; in other words, an SCP is a peak that is chemically pure, such as would be obtained on chromatographing a single compound. The attribute R*s is the average minimum resolution, which measures the average smallest interval between adjacent SCPs that are separated. R*s is not a free parameter, but it depends on the type of interpeak statistics function (for example, for random spacing of SCPs or ordered, such as fractal spacing of SCPs), the amount of peak overlap, and the distribution of SCP heights (2).
The attribute R*s differs from the traditional resolution Rs, which is a parameter freely chosen by the researcher. The traditional resolution Rs is an important attribute of the peak capacity that is defined (2) as the number of equi-spaced SCPs that fit within a discrete time increment between t1 and tm so that

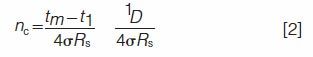
By combining equations 1 and 2, one obtains an alternate metric of peak crowding, the effective saturation αe

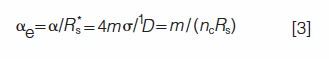
which depends only on m, σ, and 1D (3). The effective saturation is a practitioner-friendly metric for comparing peak overlap in different separations, because it is independent of R*s, which varies, as noted above, with saturation. The predictions of SOT are derived relative to α, but are more easily interpreted relative to αe.
Different approaches to SOT have been proposed, including some based on Fourier analysis (4,5) and pulse-point statistics (6). Various reviews of the different methods have been published (7,8). In this article, we consider only point-process statistics, in which the distribution of intervals between the retention times of successive SCPs is considered. For simplicity, we consider only cases where SCPs are spread more or less equally throughout the separation.
The simplest interpeak statistics function in SOT is based on Poisson statistics, which assume that SCPs are distributed across the separation space randomly. This assumption is well-founded both empirically (1,9–12) and theoretically (13,14) for a number of mixtures. This random placement of SCPs requires that the arrival times of SCPs are governed by a Poisson process based on exponential waiting times (15):


where P(t) is the probability density of finding the next SCP some time t after the last SCP. This relationship governs such random processes as radioactive decay and is called a renewal process (16) in the probability literature. The quantity λ in equation 4 is the component density or the number of components per total separation space so that λ = m/1D. The expectation (or average) value of the density P(t) is E and is equal to 1/λ. This can be generalized: E = 1D/m. Therefore, the expectation value of the density can be interpreted as the average separation space between SCPs.
The probability that the interval between two SCP centres exceeds some time t′= 4σR*s, allowing these SCPs to be resolved from each other is

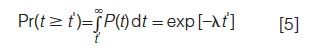
Noting that α = λt′, one ultimately finds

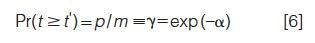
where p is the number of peaks in the chromatogram and the ratio γ = p/m is the fraction of components that are interpretable as peaks. Thus, this fraction is a simple function of the saturation α. For the appropriate choice of R*s (17,18), p is the number of visible maxima.
Another quantity of interest is the fraction of components that are singlet peaks (1):

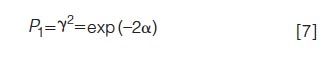
In general, the fraction Pn of components appearing in peaks containing n components (for example, for doublets, n = 2; for triplets, n = 3; and so on) is as follows (1):

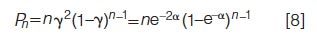
Equations 6–8 are valid for different interpeak statistics functions producing different renewal processes, as long as the ratio γ = p/m is replaced by the appropriate function of α (19).

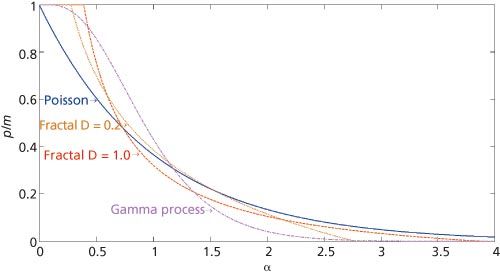
Figure 1: Plot of p/m, the fraction of peaks found in the chromatogram as a function of the saturation α for four renewal processes: Poisson (random) process, solid line; power-law (fractal) process with D = 1.0, β = 10, dashed line, D = 0.2, β = 10, dotted line; and gamma process (P = 4, as explained in reference 19), dash-dotted line. The β parameter is explained in reference 2.
Consequences of Overlap
We show the consequence of overlap using the random SCP approach developed in the equations above in Figure 1. Other renewal processes besides the (random) Poisson process, for example, two power-law (fractal) renewal processes (2) and a Γ process based on the gamma distribution (19), are also shown. While these processes differ in the assumptions of the statistical SCP spacing, the trends are apparent. The dimensionality D used in Figure 1 and below is explained in great detail in reference 2 and is a measure of the ordering of a chromatogram. At higher D values ordering increases and at low D values there are many gaps in the chromatogram.
As the saturation α increases, the fraction of components that appear as peaks decreases rapidly. In the case of SCPs that are more ordered, as found in the fractal and Γ processes, the decrease in peaks as α increases does not fall as rapidly as a random ordering, at least at low saturations. However, Figure 1 also shows that as α approaches one, ordering causes more loss of peaks than a random filling of the peak space.

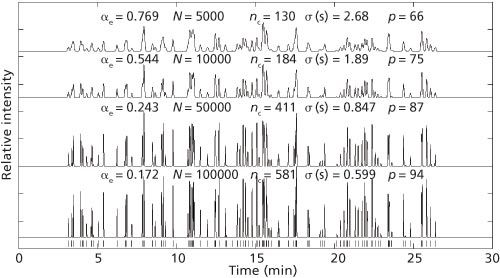
Figure 2: The effect of varying efficiency (number of plates) on the number of visible peaks. The numbers of peaks detected (p) are 94, 87, 75, and 66 for four different efficiency scenarios given 100 components. The symbols are effective saturation, αe, number of plates, N, peak capacity, nc, Gaussian standard deviation zone width in s, σ, and the number of visible peaks, p.
The consequences of overlap are shown in a complementary way in Figure 2, where we use synthetic chromatograms comprising sums of Gaussian peaks that are distributed randomly throughout the retention time range and have uniformly random heights. The zone (or SCP) standard deviation used here is obtained from the following well-known equation (20):


so that given a number N of theoretical plates and a retention time t the zone standard deviation σ is determined. For Figure 2, the retention time in equation 9 is that of the first retained SCP, and a model of constant zone width is assumed, which is approximated in temperatureâprogrammed gas chromatography (GC) and gradientâelution liquid chromatography (LC).
The four chromatograms in Figure 2 vary in efficiency (number of plates), and this is reflected in the Gaussian zone standard deviation, σ, which in turn affects αe and nc (as calculated from equations 2 and 3, with Rs = 1). The number of components, m, is 100.
The retention times are represented at the bottom of Figure 2 by stick locations that show what the chromatogram would look like, except for the distortion of peak heights, if the peaks were infinitely narrow, that is, if σ = 0 and hence αe = 0 and nc = ∞.
As can be seen from Figure 2, at high efficiency with N = 100,000 plates, 94 peaks are present. This number drops off to 87 when the plate count is reduced to 50,000. At 10,000 plates, 75 peaks are present and at 5000 plates only 66 peaks are present. Only a fraction of these peaks are singlets; a good number of these are fused doublets and triplets (and even more complex multiplets). Hence, as efficiency is reduced, as measured by increases of αe, the number of peaks present drops monotonically.
The consequences of this phenomenon are well known. Peak fusion interferes with proper quantitation. It also interferes with the identification of specific components. Often times this can be aided with mass spectrometry (MS) detection. However, this is not always the case as peak fusion can lower ionization efficiency, and mass spectrometry often cannot distinguish between closely related compounds with the same molecular weight (and hence the same parent ion).
Chromatography is particularly problematic for samples of biological origin because of the multiplicity of forms, called isoforms. These isoforms are closely related in structure (but are not the same) yet may have different chromatographic retention. In addition, many biological molecules have dynamic structures so that chromatographic retention occurs with a multiplicity of different molecular conformations, all of which lead to zone broadening and a lowering of the overall effective efficiency. These effects reduce the apparent efficiency of the chromatographic process and cause an artificial increase in α, making chromatographic separation more difficult for biomolecules than in the case of small molecules. This is why biomolecules are often denatured before analysis in the hopes of minimizing the conformational shifts during the separation process.
Use of the SOT as a Ratio
Another useful view of SOT is to express equation 6 as a ratio. In this way, we can estimate what the gain or loss of peaks will be by changing efficiencies at constant sample, constant selectivity, and constant relative solvent programme.
A common shortcoming in SOT calculations is the failure to distinguish between the freely chosen traditional resolution Rs and the average minimum resolution Rs. The assumption that they are the same introduces error, and the distinction must be kept in rigorous work. However, it is convenient to identify them to simplify matters and evaluate trends. We do so here for simplicity’s sake, but the results obtained must be interpreted as only guidelines.
Consider a case in which the duration 1D of two separations is the same but the SCP standard deviations therein are different. Evaluating the ratio of equation 6 with fixed m (constant sample component number), one finds, with the subscripts denoting two different columns:

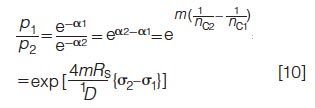
where α1 and α2 are two different saturations, nc1 and nc2 are two different peak capacities, and σ1 and σ2 are two different SCP standard deviations.
Using equation 10 and H = L/N, where H is the plate height, L is the column length, and the nondimensional retention parameter k′ = (t/t0)–1, with t0 equaling the void time, one can show that

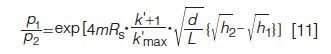
where d is the particle diameter, k′max is the maximum k′ used in the analysis, and h is the nondimensional plate height, H/d. As an example in LC, consider the chromatographic values of Rs = 1, k′ = 5, k′max = 20, L = 15 cm, d = 2.7 µm, h2 = 1.5, and h1 = 1.0. For these parameters, the ratio in equation 11 for a 200 component mixture (m = 200) is equal to 1.25, indicating that 25% more peaks would appear in a chromatogram using a column that was extremely high in efficiency where h = 1.0, as compared to a more conventional very high performance column, for example a core–shell particle where h = 1.5. This number suggests that the pursuit of even higher performance column technology is a most desirable goal in increasing the number of detectable peaks. Furthermore, it is known that even if this level of performance is not warranted, the speed of separation can be increased when high efficiency column technology is utilized.
For situations where zones are ordered, using fractal statistics, the ratio approach is powerful. It can be shown (2) that under limiting conditions the ratio of the number of peaks found is related to the two plate counts, N1 and N2, and the fractal dimension D, such that


Multidimensional Separation by k Columns
The results presented earlier show that the limited separation space of one column, even those of very high efficiency, still has limited separation capabilities. Of historic interest is the use of multiple columns to increase the likelihood that a given compound is separated by at least one column. The probability of success was first addressed by Connors (21) and subsequently reexamined (22). For k separations (that is, columns) of the same mixture, with the separations having the same saturation but independent separation mechanisms, the probability s/m that a component appears as a singlet peak on at least one column is

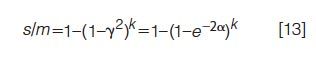
where the last equality applies to a Poisson distribution of SCPs. Figure 3 is a graph of the final expression in equation 13 for k values between 1 and 5. The k = 1 graph represents separation by a single column. As k increases, the likelihood of separation increases. For a saturation α equal to 1, corresponding to a separation of moderate difficulty, the likelihood that a component of interest is resolved as a singlet peak on a single column (k = 1) is only 14%. However, this number increases to 25%, then 35%, then 44%, and finally 52% as the number of columns is increased from two, to three, to four, and finally to five.

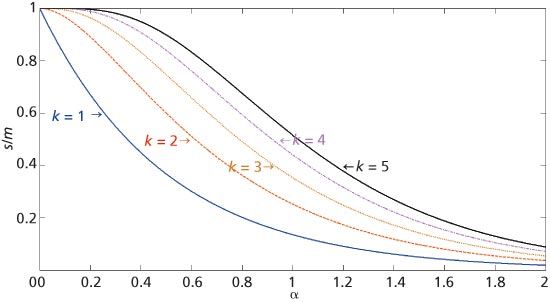
Figure 3: Graph of the probability that a given component appears as a singlet peak, s/m, versus the saturation of k independent columns.
Other types of multidimensional separations (k ⩾ 2) can be considered. A classic method is column switching, in which a subsection of the entire chromatogram is transferred to another column. Two-dimensional chromatography (k = 2) attempts to increase the peak capacity by providing a separation area rather than a line, and this is needed for very complex mixtures. A few cases of separations in higher dimensions (for example, k = 3) have been reported. SOTs have been developed for all of these methods (23–26).
Conclusions
SOT started in the early 1980s, and a glance at the references below shows that many were published long ago. What is the relevance of SOT today? The hope of early researchers that SOT could be used routinely to estimate the number of components in mixtures is largely unfulfilled. The reason is that too many unknowns exist in real chromatograms, perhaps most importantly the type of interpeak statistics function (and its possible variation over the separation) governing the spacing between SCPs. The large variation of SOT predictions for different functions is shown in Figure 1. Various functions have been proposed over the years (1,2,4,19), with assessments based on the quality of their description of experimental data. Nevertheless, an infinite number of such functions can be proposed (based on known or empirical statistical laws), and unless one knows on a physicochemical basis what functions are favoured for a given mixture and column, one is left with uncertainty.
However, SOT does serve two purposes. First, it provides the basis for semiquantitative to quantitative predictions of the expected outcome of the completeness of a separation in a chromatogram, and how much effort is required to improve that chromatogram. This is especially true for a single column, where the validity of the assumption of Poisson statistics is often justified. Several such predictions were presented here. Second, theory can be used to model attributes of interest in chromatograms, even when the assumptions of SOT do not apply. For example, the correlation of retention times in many two-dimensional chromatograms invalidates the assumption of SCP randomness. Nevertheless, twoâdimensional SOT has been used on model systems to understand the undersampling of firstâdimension peaks in comprehensive twoâdimensional chromatography (27), the improvement of resolution therein by the use of multivariate selectivity (28), and the comparison of one- and two-dimensional chromatography (29).
SOT offers powerful, yet practical insight into the statistical mechanics of separation. As with many areas of separation science, the development of SOT is an interdisciplinary task, in this case between chemistry and applied probability theory. The initial mathematical difficulties have been overcome by a continuous refinement of SOT conducted by a multitude of authors involved in developing and refining chromatographic theory. The results and predictions are meaningful and make very practical guides to experimental methods development.
Biomedical areas of research such as the search for biomarkers, metabolomics analysis, and proteomics research, all deal with saturation issues in chromatography. The consequences of saturation include an undeniable loss in unique identification in single channel detectors. The instrumental development of the chemical analysis process requires coupling high resolution columns, perhaps even multiple separation stages, together with multichannel detectors such as mass spectrometers and multiple MS stages. Sometimes zones can be resolved with unique ion identification schemes, and sometimes zones have mixtures that are not resolvable by MS. This coupling and its refinement towards reaching reliable molecular identification needs to be understood quantitatively in the context of chromatography by the extension of SOT.
References
- J.M. Davis and J.C. Giddings, Anal. Chem. 55, 418–424 (1983).
- M.R. Schure and J.M. Davis, J. Chromatogr. A1218, 9297–9306 (2011).
- J.M. Davis and P.W. Carr, Anal. Chem. 81, 1198–1207 (2009).
- A. Felinger, L. Pasti, and F. Dondi, Anal. Chem.62, 1846–1853 (1990).
- M.C. Pietrogrande, M.G. Zampolli, and F. Dondi, Anal. Chem.78, 2579–2592 (2006).
- F. Dondi, A. Bassi, A. Cavazzini, and M.C. Pietrogrande, Anal. Chem.70, 766–773 (1998).
- M.C. Pietrogrande, A. Cavazzini, and F. Dondi, Rev. Anal. Chem.19, 123–156 (2000).
- A. Felinger and M.C. Pietrogrande, Anal. Chem. 73, 619A–626A (2001).
- D.P. Herman, M.-F. Gonnord, and G. Guiochon, Anal. Chem.56, 995–1003 (1984).
- M. Martin, D.P. Herman, and G. Guiochon, Anal. Chem. 58, 2000–2007 (1986).
- C. Samuel and J.M. Davis, J. Chromatogr. A842, 65–77 (1999).
- C. Samuel and J.M. Davis, J. Microcol. Sep.12, 211–225 (2000).
- A. Felinger, Anal. Chem. 67, 2078–2087 (1995).
- J.M. Davis, M. Pompe, and C. Samuel, Anal. Chem. 72, 5700–5713 (2000).
- J.F.C. Kingman, Poisson Processes (Oxford University Press, 2002).
- D.R. Cox, Renewal Theory (Methuen & Co. 1967).
- A. Felinger, Anal. Chem.69, 2976–2979 (1997).
- J.M. Davis, Anal. Chem. 69, 3796–3805 (1997).
- M.C. Pietrogrande, F. Dondi, A. Felinger, and J.M. Davis, Chemom. Intell. Lab. Sys.28, 239–258 (1995).
- J.C. Giddings, Unified Separation Science (Wiley, 1991).
- K.A. Connors, Anal. Chem. 46, 53–58 (1974).
- J.M. Davis and L.M. Blumberg, J. Chromatogr. A 1096, 28–39 (2005).
- J.M. Davis, Anal. Chem. 65, 2014–2023 (1993).
- M. Martin, Fresenius’J. Anal. Chem.352, 625–632 (1995).
- C. Samuel and J.M. Davis, Anal. Chem.74, 2293–2305 (2002).
- S. Liu and J.M. Davis, J. Chromatogr. A1126, 244–256 (2006).
- J.M. Davis, D.R. Stoll, and P.W. Carr, Anal. Chem.80, 461–473 (2008).
- J.M. Davis, S.C. Rutan, and P.W. Carr, J. Chromatogr. A 1218, 5819–5828 (2011).
- J.M. Davis, Talanta83, 1068–1073 (2011).
Mark R. Schure is with Kroungold Analytical, Inc. in Blue Bell, Pennsylvania, USA. Joe M. Davis is with the Department of Chemistry and Biochemistry at Southern Illinois University at Carbondale in Carbondale, Illinois, USA. Direct correspondence to: Mark.Schure@gmail.com







Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.






