Separation Science in Drug Development, Part 3: Analytical Development
LCGC Europe
The third instalment in this series provides an overview of modern practices of separation science in small-molecule drug development. It highlights approaches in high performance liquid chromatography (HPLC) method development and physical or chemical characterization to support process chemistry and formulation development, and for assessment or control of clinical trial materials. The roles of separation scientists in analytical development and salient chromatographic methodology trends are also discussed.
Michael W. Dong, Perspectives in Modern HPLC Editor
The third instalment in this series provides an overview of modern practices of separation science in small-molecule drug development. It highlights approaches in high performance liquid chromatography (HPLC) method development and physical or chemical characterization to support process chemistry and formulation development, and for assessment or control of clinical trial materials. The roles of separation scientists in analytical development and salient chromatographic methodology trends are also discussed.
The pharmaceutical industry is a major employer of analytical chemists, particularly separation scientists skilled in high performance liquid chromatography (HPLC) (1,2). The third and fourth instalments in this series of articles describe separation science methodologies and processes in analytical development and quality control for new drug product development. Following the synthesis of a new chemical entity (NCE) with acceptable physicochemical properties as well as preliminary safety and efficacy profiles (3), serious efforts in analytical chemistry are required before its nomination as a clinical development candidate. This milestone is marked by a commitment to conduct a good laboratory practice (GLP) toxicology (tox) study representing a significant financial investment from the company (1). At this point, the NCE should be reasonably well characterized from a physicalâchemical point of view, and the synthetic route well defined to allow process development of a scaleâup synthetic route. One of the first tasks of the analytical chemist is the development of a stabilityâindicating HPLC assay to investigate the purity and stability of the drug substance (DS) (4,5). The next step is the selection of a preferred solid-state form of the drug substance (free acid–base form or salt, amorphous–crystallinity, polymorphs) based on their stability, solubility, and the intended route of administration (for example, oral or parenteral) (6). Planning for the regulatory filing needed to conduct clinical trials (such as investigational new drug [IND] applications in the United States or clinical trial applications [CTA] in the European Union) then begins in earnest. Since clinical trial materials (CTM), which consist of both drug substances and drug products (DP), used in the clinics are typically produced under good manufacturing practice (GMP) regulatory guidelines, the initial quality specifications are proposed and quality control (QC) methodologies are established for the CTM (7).


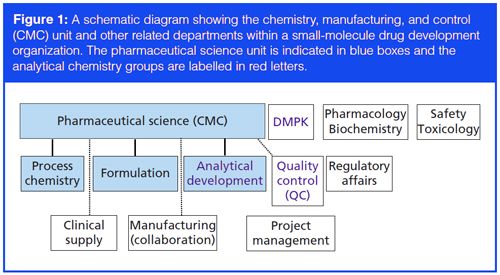
A schematic diagram of a typical organization structure in the pharmaceutical company for technical development, generally categorized as chemistry, manufacturing, and control (CMC) activities described in the regulatory dossier, are shown in Figure 1. The pharmaceutical science unit is generally responsible for the CMC functions and has three typical subgroups:
- A process chemistry group to scale up the synthetic route of the drug substances.
- A formulation or pharmaceutics group for formulation development and production of drug products for the clinic.
- An analytical development group to develop methodologies to characterize and assess the quality of DS and DP lots and to support process development.
The pharmaceutical science unit works collaboratively with various departments in the organization, such as drug metabolism and pharmacokinetics (DMPK), safety–toxicology, pharmacology–biochemistry, quality assurance, regulatory affairs (RA), project management, and clinical supplies (1). Its responsibilities are the technical and process development of all CTM and the documentation of the CMC sections in regulatory filings before new drug application (NDA) registration. A brief synopsis of their respective functions is available elsewhere (1).
This instalment provides a brief overview of the analytical development activities for new smallâmolecule drug candidates (1,2,4) with a focus on the applications of modern separation science methodologies. The discussion is limited to smallâmolecule drugs because the methodologies and quality requirements are considerably different from those for large-molecule biologics (8). Not surprisingly, HPLC is the primary methodology (2) and is complemented by gas chromatography (GC), ion chromatography (IC), supercritical fluid chromatography (SFC), and other analytical techniques for specific applications. The roles played by analytical chemists in method development, impurities identification, and sample analysis are described along with prominent trends of separation science methodologies as they manifest themselves in these activities.
The Role of Analytical Chemists in Analytical Development and Quality Control
Table 1 summarizes the roles of analytical chemists in analytical development and quality control in smallâmolecule drug development (2,6). The analytical development (AD) and QC groups are usually separate in the organization but can be merged in smaller companies.


Shortly before the nomination of the new drug candidate, a multidisciplinary CMC team is formed consisting of representatives from process chemistry, formulation development, analytical chemistry, quality assurance, regulatory affairs, project management, and other groups as necessary. An immediate goal of the CMC team is to supply sufficient drug substance for GLP tox studies. The typical first tasks for the analytical chemist are to fully characterize the NCE and the development of a stabilityâindicating HPLC method (4,6). The next steps are to provide technical support for process chemistry and pharmaceutics. Note that these development activities can be conducted by internal staff or partially or completely outsourced to contract manufacturing organizations (CMO). Other tasks may include conducting preliminary stability studies and the identification of key impurities and degradation products.
The manufacturing and testing of clinical trial materials follow strict current GMP (cGMP) guidelines. Here, QC chemists are responsible for method validation and release testing of all batches of DS and DP. The first clinical batches are put on formal stability studies to establish shelf life, packaging, shipping, and storage conditions. The QC chemist establishes stageâappropriate specifications of CTM and is responsible for the technical contents of CMC sections in regulatory filings, which include details on physicochemical properties, analytical methods and validation, impurities identification, batch analysis, and stability data. The QC and regulatory functions of analytical chemists will be discussed in the next instalment of this series.
Characterization Techniques for Drug Substances and Drug Products
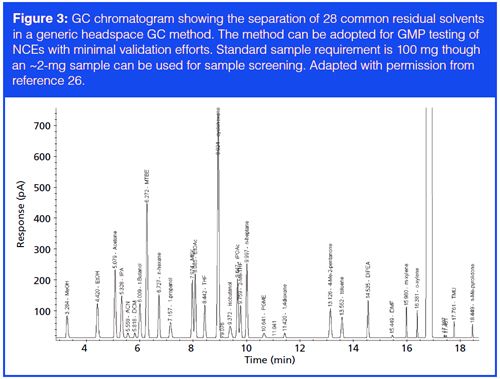
Table 2 summarizes the primary characterization and QC techniques for DS and DP, which include chromatography, spectroscopy, thermal analysis, titration, and other analytical procedures (9). This typical set of analytical testing provides a scientific understanding of the physicochemical properties that may impact critical quality attributes (CQAs, which are defined as those important attributes affecting the safety and efficacy of the drug) of the CTM in process and product development (1,6). Note that chromatographic techniques (those underlined in Table 2), particularly HPLC, provide the predominant methodologies for purity, potency, and DP performance (dissolution) determinations. CQAs are captured in the acceptance criteria (specifications), which must be confirmed by QC testing to be within specifications for every batch of DS and DP before they can be released for clinical use (that is, release testing). In contrast to analytical methodologies used in high-throughput characterization, as described in instalment two of this series (10), with their focus on fast turnaround time, QC procedures have high accuracy and reliability and must be qualified before use for release testing (4).

In the next section, modern practices and trends of separation science methodologies in several selected areas are described: HPLC method development; chiral separations and analysis of molecules with multiple chiral centres; the use of generic platform multicomponent methods (HPLC, GC, and IC); impurity identification using LC coupled to mass spectrometry (MS); highâresolution HPLC analysis of complex drug products with multiple active pharmaceutical ingredients (APIs) by ultrahighâpressure LC (UHPLC); and analytical strategies for genotoxic impurities (GTI).
HPLC Method Development Approaches and Trends
There are numerous books, articles, and training courses devoted to HPLC method development (2–4,11), so only highlights of general approaches and new trends are described here. The first task in analytical development is the development of a stabilityâindicating HPLC method, which is defined as a validated analytical procedure that accurately measures active ingredients free from potential interferences such as degradation products, process impurities, excipients, and other potential impurities (12,13). This method is used to establish the impurity profile of the drug substance lot used in the GLP tox study and in subsequent clinical or stability lots. With few exceptions, a single reversed-phase LC method is used for both release testing and during stability studies (4,13).
The traditional approach in HPLC method development was described in detail by Snyder and colleagues (11) and others (4,5). It involves the following steps:
- Defining method and separation goals
- Gathering sample and analyte information
- Initial method development - “scouting” runs and getting the first chromatogram
- Method fine-tuning or optimization
- Method validation
This traditional approach is a systematic but time-consuming process and is most appropriate for critical methods for purity assessment of important materials (for example, drug substances). Note that step 4 (method optimization) consumes the majority of time and effort in the method development process. During early drug development, numerous HPLC methods are needed for raw materials (RM) and starting materials (SM) where substantial efforts using the traditional approach may not be justified or required. For rapid method development, a practical threeâpronged approach for rapid HPLC method development was proposed with three method types of increasing complexity (14):
- Fast LC isocratic potency and performance methods
- Generic broad gradient methods for purity assays of less-complex materials
- Multisegment gradient methods for NCEs and complex samples
The characteristics, advantages, and limitations of each method type are described in more details elsewhere (14). Recognizing the method characteristics associated with different assay purposes or sample types will expedite the method development process. This approach is most useful during early drug development when quick response time is important.


Modern trends in HPLC method development are summarized in Table 3 and have been discussed elsewhere (4,5). Several significant trends (those underlined) deserve a brief comment here. First is the increased use of MS-compatible methods to facilitate peak identification and peak tracking even though ultaviolet (UV) detection continues to provide quantitation data in most cases. A potential downside for MS-compatible methods is lower sensitivity in the far UV region (such as <220 nm) because of the absorbance of volatile mobile-phase additives such as formic acid, acetic acid, or ammonium salts of carboxylic acids (4). Secondly, UHPLC with short columns and small-particle stationary phases reduces analysis time and is increasingly used in method development and sample analysis (15,16). The combination of lower system dwell volumes with UHPLC, shorter gradient times, and high-efficiency columns shortens the time for column screening to less than an hour with similar time savings for mobile phase and gradient optimization. A disadvantage, however, is the potential need for converting the UHPLC methods to HPLC-compatible conditions using longer columns packed with larger particles (17). This conversion may be required for global manufacturing of the CTM and associated analytical testing around the world because some laboratories may not have UHPLC capabilities. Lastly, automated method development systems with column and mobile-phase screening capabilities, built-in data analysis, and method optimization functionalities are becoming more prevalent and are particularly useful for the development of robust HPLC methods for complex molecules or drug products (18).
Chiral Separations and Method Development Strategy for Molecules with Multiple Chiral Centres
Development of NCEs with high chemical and chiral purity is a regulatory expectation in new drug development (1,6,19). Today, many drugs are chiral and it is not unusual to have an NCE with more than one stereogenic centre. The most effective approach for the determination of enantiomers is chiral LC (19,20) or chiral SFC (21) using polysaccharideâbased columns under normal-phase LC conditions. Unlike reversed-phase LC, whose separation mechanism is highly predictable, development of chiral LC methods is reliant on column and mobile-phase screening experiments (19–21). Chiral SFC has the advantage of analysis speed and sample recovery versus LC, and is preferred for column and mobile screening and for preparative applications (3).
For chemical development of multichiral molecules with multiple chiral centres, significantly higher numbers of analytical methods capable of separating all stereoisomers (enantiomers and diastereomers) must be developed rapidly to assess and control the stereochemistry of raw materials (RM), intermediates, and the APIs. Paradoxically, achiral reversedâphase LC methods, used to assess the overall chemical purity of the API, can usually be developed to monitor the content of multiple diastereomers in a single run (14,22). In many cases, these achiral reversedâphase LC methods often become the primary stability-indicating QC methods. Chiral methods for the determination of the enantiomers in the drug substance are desirable but may not be required if control of the stereochemistry of the starting materials and the lack of epimerization during the synthetic process can be demonstrated.
The Increased Use of Generic Multicomponent Platform Methods
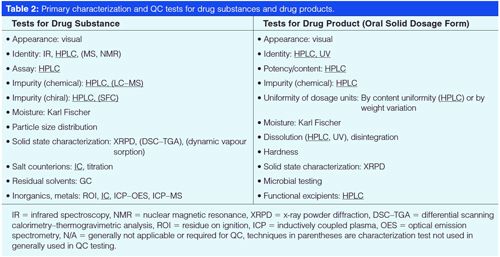
While each NCE may require customized analytical procedures, generic platform methodologies can frequently be applied successfully with little or no modifications. For instance, generic broad gradient HPLC methods have been used effectively for high-throughput screening and in-process control testing (3,14). Recently, a universal 10-min HPLC–UV cleaning verification method was published that works well for multiple drug candidates (23). The advent of UHPLC and columns packed with sub-2-µm stationary phases has extended the speed and effectiveness of this approach (15,18). The availability of columns packed with sub-2- or sub-3-µm superficially porous particles (SPP) (24), which increases efficiency by 20–30% versus their totally porous counterparts, further enhances the versatility of these generic methodologies. Figure 2 shows the separation of 10 peaks in a mixture of 12 NCEs in 2 min (Figure 2[a]) using a modernized generic method on a 5-cm long SPP column. Note that all 12 components can be separated in 5 min (Figure 2[b]) with excellent peak shapes and sensitivity by quick modifications of the gradient conditions (Figure 2[a]) (25). This type of generic gradient method using optimized columns and gradient conditions may also prove useful as initial purity method conditions for many raw materials, starting materials, intermediates, or drug substances (25).

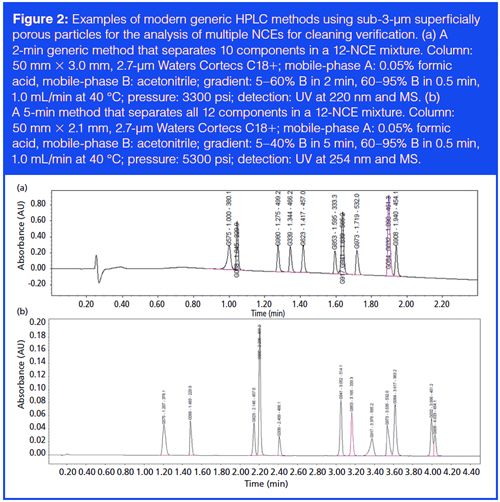
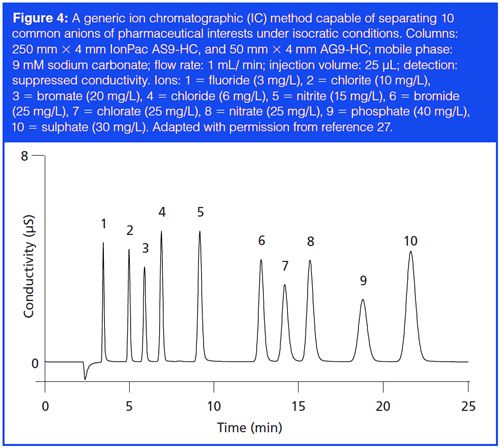
Other generic methods worth mentioning are the analysis of residual solvents by headspace GC (26) and the determination of pharmaceutical counterions by IC (27) or mixed-mode chromatography with charged aerosol detection (CAD) (28). Chromatograms and method details of the two example generic methods by GC and IC are shown in Figures 3 and 4, respectively. The readers are referred to the original articles for more details.


Impurities Identification by LC–MS
Impurity identification may occur at various phases by the analytical chemist often in conjunction with an MS and nuclear magnetic resonance (NMR) expert. Forced degradation studies are typically conducted on the NCE during HPLC method development under a set of stress conditions (acid, base, oxidative, heat, light, moisture) (29). LC–MS analysis of these forced degradation samples can provide identification of degradation products and yield information on the primary degradative pathways. For instance, a singleâquadrupole MS system can provide protonated molecular ion (M+H) data for the preliminary identification of isomers, dimers, other impurities, and oxidative or hydrolytic degradation products. Furthermore, newer lowâcost, compact single-quadrupole MS systems may allow easier adoption of single-quadrupole MS for method development and sample characterization by general analytical chemists without extensive training in MS (30).
Major impurities and degradation products in the GLP drug substance tox lot and from initial stability studies should be identified by LC–MS and NMR in accordance with the International Conference on Harmonization’s (ICH) guideline Q3A (R3) (12,31). High-resolution MS is often used to provide moreâdefinitive information. The levels of impurities in the tox lot are important because they are considered to be biologically qualified in a tox study up to a certain level (typically called the “no-observed-adverse-effect-level” or NOAEL) (1). They are used to justify specifications for specified impurities for the clinical lots.
Analysis of Complex Drug Products with Multiple APIs Using UHPLC
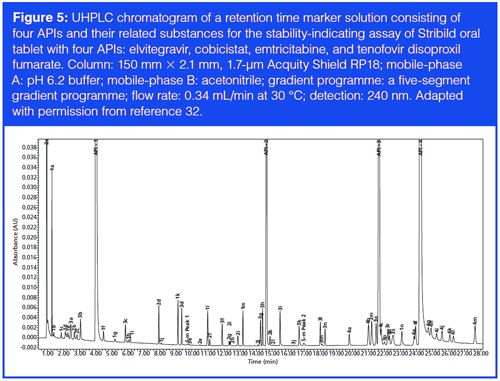
Another significant benefit of UHPLC is its high resolving power for the analysis of very complex samples, which is difficult to achieve with conventional HPLC because it has a theoretical maximum peak capacity of ~200 for routine testing (6). The superb resolving power of UHPLC is demonstrated in the analysis of a combinational drug product with four APIs (stribild oral tablet with elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate) shown in Figure 5. This method was developed and optimized using an automated method development system and was validated for release testing and for use during stability studies (32).

Genotoxic Impurities
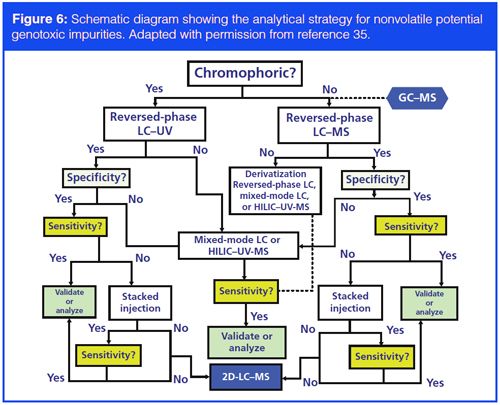
A topic of increasing concern to the regulators is the presence of genotoxic impurities (GTI) and potential genotoxic impurities (PGI) in pharmaceuticals including CTM (33), also termed potential mutagenic impurities in the recently released ICH M7 guidelines (34). PGIs can be residual reagents or side-products, and they can be reactive and have diversified structures. Since these GTIs and PGIs may need to be controlled at parts-per-million levels, very sensitive and selective methodologies are required. The development of analytical procedures for GTI and PGI can be extremely challenging and resource-intensive. While generic methods using LC–UV, LC–MS, or LC–MS–MS may be effective at times, additional tactics with specific sample extraction or derivatization are often needed in practice. An analytical strategy for PGI has been proposed (35) and is included as a reference guide in Figure 6. Most laboratories develop methods for PGI to demonstrate the impurity-purging capability of the chemical process during development so that QC testing is not required in the final product (35).

Significant Trends of Separation Science Methodologies
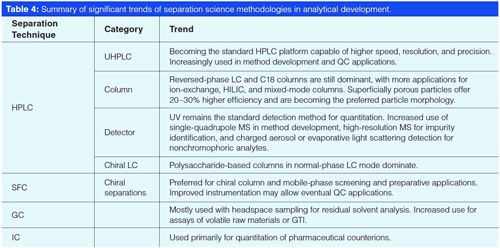
Table 4 summarizes significant trends of separation science methodologies in analytical development. The adoption of UHPLC in global analytical testing remains an issue and will face challenges until UHPLC equipment is universally available in all laboratories. Reversedâphase LC using C18 materials still predominates with trends towards smaller particle size (subâ3â and subâ2âµm) and superficially porous particles. UV detection remains the detection method of choice in pharmaceutical analysis supplemented by MS for peak identification or trace analysis, and CAD or evaporative light scattering detection (ELSD) for nonchromophoric compounds. Chiral LC is increasingly used for process analysis and quality control of chiral drugs with higher adoption of SFC in column and mobile phase screening. GC applications are limited mostly for residual solvent determination and the analysis of volatile raw materials or genotoxic impurities. Ion chromatography is primarily used for the determination of pharmaceutical counterions.

Summary and Conclusion
Separation science methodologies in the characterization of drug substances and drug products, method development, chiral separations, impurities and degradation products identification, and analyzing complex samples were described here. The examples shown include generic methods for cleaning verification, residual solvents, and pharmaceutical counterions, and the analysis of drug products with multiple APIs. Significant trends in analytical development include the increased use of UHPLC, SFC, MS, and automated systems in method development and high-resolution MS in impurity identification, as well as the emerging predominance of superficially porous particles as the preferred column packing morphology. The analysis of molecules multiple chiral centres and genotoxic impurities represents both a challenge and an opportunity for the separation scientist.
Acknowledgements
The author would like to thank Drs. Dawen Kou, Christina Gu, and Mengling Wong of Genentech; Dr. Jessica Lin of Gilead Sciences; Kim Huynh-Ba of Pharmalytik; Dr. Todd Maloney of Eli Lilly and Company; Dr. Raphael Ornaf of Vertex Pharmaceuticals; and Dr. Szabolcs Fekete of the University Geneva, for their editorial input and suggestions.
References
- Drug Discovery and Development: Technology in Transition, 2nd edition, R.G. Hill and H.P. Rang, Eds. (Churchill Livingston, Edinburgh, Scotland, 2012).
- HPLC for Pharmaceutical Scientists, Y.V. Kazakevich and R. LoBrutto, Eds. (Wiley, Hoboken, New Jersey, USA, 2007), Chapters 7, 14, 15, and 22.
- M. Wong, B. Murphy, J. Pease, and M.W. Dong, LCGC Europe28(6), 343–352 (2015).
- M.W. Dong, Modern HPLC for Practicing Scientists (Wiley, Hoboken, New Jersey, USA, 2006), Chapters 2, 4, and 8.
- H.T. Rasmussen, W. Li, D. Redlich, and M.I. Jimidar, in Handbook of Pharmaceutical Analysis by HPLC, S. Ahuja and M.W. Dong, Eds. (Elsevier/Academic Press, Amsterdam, the Netherlands, 2005), Chapter 6.
- M.W. Dong, “Drug Discovery and Development Processes,” presented at Pittcon 2015, New Orleans, Louisiana, USA, 2015.
- Current Good Manufacturing Practice for Finished Pharmaceuticals, in Code of Federal Regulations Title 21 Part 211 (U.S. Government Printing Office, Washington, DC, USA, 2009).
- T. Zhang, C. Quan, and M.W. Dong, LCGC Europe27(11), 595–609 (2014).
- D.A. Skoog, F.J. Holler, and S.R. Crouch, Principles of Instrumental Analysis, 6th ed. (Brooks Cole, Independence, Kentucky, USA, 2006).
- B. Lin, J. Pease, and M.W. Dong, LCGC Europe28(8), 455–463 (2015).
- L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid chromatography, 3rd Ed. (John Wiley & Sons, Hoboken, New Jersey, USA, 2010).
- International Conference on Harmonization, Q3A (R2): Impurities in new drug substances (ICH, Geneva, Switzerland, 2006).
- M.W. Dong, LCGC Europe26(6), 393–309 (2013).
- M.W. Dong, LCGC Europe26(8), 455–477 (2013).
- M.W. Dong, LCGC North Am.25(7), 656–666 (2007).
- M.W. Dong and K. Zhang, Trend. in Anal. Chem.63, 21–30 (2014),
- M.W. Dong, LCGC Europe26(11), 637–649 (2013).
- D. Guillarme and M.W. Dong, Amer. Pharm. Rev.16(4), 36–43 (2013).
- Y. Vander Heyden, D. Mangelings, N. Matthijs, and C. Perrin, Chiral Separations, in Handbook of Pharmaceutical Analysis by HPLC, S. Ahuja and M.W. Dong, Eds. (Elsevier/Academic Press, Amsterdam, The Netherlands, 2005), Chapter 18.
- C. Hamman, M. Wong, M. Hayes, and P. Gibbons, J. Chromatogr. A1218(22), 3629–36 (2011).
- Supercritical Fluid Chromatography: Advances and Applications in Pharmaceutical Analysis, G.K. Webster, Ed. (CRC Press, Taylor and Francis, Boca Raton, Florida, USA, 2014).
- M.W. Dong, M. Goel, and K. Zhang, “HPLC Method Development for New Drug Candidates with Multiple Chiral Centers,” presented at Pittcon 2015, New Orleans, Louisiana, USA, 2015.
- M.W. Dong, E.X. Zhao, D.T. Yazzie, C.C. Gu, and J.D. Pellett, Amer. Pharm. Rev.15(6), 10–17 (2012).
- S. Fekete, D. Guillarme, and M.W. Dong, LCGC North Am.32(6), 420–433 (2014).
- M.W. Dong, “Generic HPLC Method(s) in Pharmaceutical Analysis,” presented at Waters Acquity Users’ Meeting, Foster City, California, USA, 2014.
- L. Dai, A.C. Quiroga, K. Zhang, H.B. Runes, D.T. Yazzie, K. Mistry, N.P. Chetwyn, and M.W. Dong, LCGC North Am.28(1), 54–66 (2010).
- C. Pohl, in Handbook of Pharmaceutical Analysis by HPLC, S. Ahuja and M.W. Dong, Eds. (Elsevier/Academic Press, Amsterdam, the Netherlands, 2005), Chapter 8.
- K. Zhang, L. Dai, and N. Chetwyn, J. Chromatogr. A1217, 5776–5784 (2010).
- S.W. Baertshi, Pharmaceutical Stress Testing (Taylor and Francis, New York, New York, USA, 2005).
- M.W. Dong, “Evaluation of a Low-Cost Mass Spectrometer,” presented at Pittcon 2015, New Orleans, Louisiana, USA, 2015.
- Handbook of Stability Testing in Pharmaceutical Development, K. HuynhâBa, Ed. (Springer, New York, New York, USA, 2009).
- M.W. Dong, D. Guillarme, S. Fekete, R. Rangelova, J. Richards, D. Prudhomme, and N.P. Chetwyn, LCGC North Am.32(11), 868–876 (2014).
- Genotoxic Impurities: Strategies for Identification and Control, A. Teasdale, Ed. (Wiley, Hoboken, New Jersey, USA, 2011).
- International Conference on Harmonization, ICH M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk (ICH, Geneva, Switzerland, 2014).
- C.J. Venkatramani and M.A. Al-Sayah. Amer. Pharm. Rev.17(5), 64–70 (2014).
Michael W. Dong is a principal in MWD Consulting focusing on training and consulting services in HPLC, UHPLC, pharmaceutical analysis, and drug quality. He was formerly Senior Scientist at Genentech, Small Molecule Analytical Chemistry and QC, Research Director at Synomics Pharma, Research Fellow at Purdue Pharma, and Senior Staff Scientist at Applied Biosystems/PerkinElmer. He holds a PhD in Analytical Chemistry from City University of New York, and a certificate in Biotechnology at University of California, Santa Cruz. He has more than 100 publications and a best-seller in chromatography. He is an editorial advisory board member of LCGC North America. Direct correspondence about this column should be e-mailed to the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com















Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



