Sample Treatment in Routine Analysis: A Case Study: Sulphonamides in Liver
LCGC Europe
Currently, sample treatment is still the bottleneck in the development of analytical methods to analyze complex samples, especially for routine analysis where high-tech instruments are not always available. Research on the evaluation of different sample treatments is needed to achieve the sensitivity and selectivity required. This article presents a case study related to the determination of sulphonamides in liver. Different extraction strategies, including solid-liquid extraction (SLE), ultrasound-assisted extraction (UAE), and solid-phase extraction (SPE), were evaluated to produce a simple and effective extraction method.
Currently, sample treatment is still the bottleneck in the development of analytical methods to analyze complex samples, especially for routine analysis where high-tech instruments are not always available. Research on the evaluation of different sample treatments is needed to achieve the sensitivity and selectivity required. This article presents a case study related to the determination of sulphonamides in liver. Different extraction strategies, including solid-liquid extraction (SLE), ultrasound-assisted extraction (UAE), and solid-phase extraction (SPE), were evaluated to produce a simple and effective extraction method.



One of the current roles of analytical scientists is to determine low concentrations of different compounds in complex matrices. The ability to quantify low concentrations of compounds in an analytical method depends on the instrumental technique as well as the sample preparation. In recent years, the sensitivity of detection techniques has been improved by the development of mass spectrometry (MS)âbased detectors (1). However, sample preparation still plays a crucial role in the development of an analytical procedure and is considered the most challenging and variable step (2,3). The choice of sample treatment depends largely on the matrix type, the method, and the physicochemical features of the analytes. The typical steps involved in sample preparation include sampling, extraction, clean-up, and concentration, followed by the final analysis (4). In addition, the development of cost-effective analytical methods is a crucial factor for achieving versatile and reliable results while largely avoiding the use of many high-tech instrumentation resources that lead to an increase in the cost of the analysis.
An ideal extraction technique should achieve selective total extraction of the target compounds (complete recoveries) while limiting the extraction of matrix impurities. In addition, compromises are needed to ensure the technique is fast, easy, and cheap as well as reducing organic solvent consumption. For solid samples, among the most used extraction techniques are pressurized liquid extraction (PLE), microwave-assisted extraction (MAE), and supercritical fluid extraction (SFE). However, these techniques are all instrument-based and as such have higher associated costs and not all laboratories have these instruments (5,6).
Traditional solid-liquid extraction (SLE) techniques are thought to be timeâconsuming, lack efficiency in extracting target compounds, and require large volumes of solvents. Nevertheless, this technique is still widely used because of its simplicity and it does not require expensive equipment (5,6). As a result of these features, this is often the only affordable technique available in laboratories where routine analytical methods are developed.
Developments in classical SLE, where shaking by hand usually ensures the partitioning of the analytes between the solid matrix and the organic solvent, include sonication, which is generally preferred to aid this contact between the two phases and promote the extraction efficiency. This extraction technique is known as ultrasound-assisted extraction (UAE) (3,7).
Nevertheless, some studies have reported on the use of SLE, not only for its simplicity, but also because it provided the best extraction results. For instance, four common methods (solid-phase extraction [SPE], matrix solid-phase dispersion [MSPD], quick, easy, cheap, effective, rugged, and safe [QuEChERS], and SLE) were compared and the authors found that SLE followed by a clean-up step based on SPE using a hydrophilic-lipophilic balanced copolymer SPE cartridge was the most suitable approach for the simultaneous extraction of antibiotics from eggs (8). Similarly, SLE was the extraction technique of choice to extract a similar group of antibiotics from swine manures (9). In another example (10), PLE and SLE were compared for the extraction of a group of mycotoxins from rat faeces. In this study, although the extraction efficiency of PLE was higher, the authors finally developed the method using SLE because it also provided suitable results, but the method simplicity was an advantage when a large number of samples had to be analyzed.
Among the different analytical methods where sample preparation is important, the determination of veterinary residues in edible tissues, such as the liver, was focused on in this article. Antibiotics are widely administrated for therapeutic purposes, but they can also be used for growth promotion in food-producing animals. The use of antibiotics for animal growth is considered fraudulent in Europe because the residues of these compounds can persist in edible matrices (11). For this reason, the European Commission regulates their use through the establishment of maximum residue levels (MRL) in foodstuff of animal origin (12). Therefore, the presence of antibiotics in edible tissues needs to be controlled through the development of analytical methods in routine laboratories to comply with these regulations.
With regards to sample preparation when working with animal tissues, one of the main difficulties is related to the complexity associated with the high fat and protein content in these matrices (3,4). Thus, the sample preparation step is critical in the development of the analytical method. In this study, the general issue of affordable sample preparation for routine analysis is transferred to the particular case of the determination of antibiotics in liver tissue by presenting different sample preparation strategies; however, these strategies could be extended to other compounds present in other complex matrices. The only constraint that is applied is that these strategies should be simple and affordable to be widely applied in any laboratory.
Experimental
Materials and Standards: Sulfadiazine (SDZ), sulfamethoxazole (SMX), sulfamethazine (SMT), sulfapyridine (SPY), sulfathiazole (STZ), all with purity ≥95%, ammonium hydroxide solution (NH4OH), and formic acid were all obtained from Sigma-Aldrich. HPLC-grade ethyl acetate (EtAc), methanol, acetonitrile, and acetic acid (HAc) were all purchased from J.T. Baker. Ultra-pure water was obtained from a water purification system (Veolia Water). Individual stock solutions of 1000 mg/L for each sulphonamide were prepared in methanol and stored at 4 ºC. A standard working solution of 100 mg/L was prepared weekly by diluting the stock solutions with ultra-pure water and stored at 4 ºC. A hydrophilic-lipophilic balanced copolymer and a mixedâmode strong-cation exchange (SCX) polymer SPE cartridges of 150 mg format were used.
Sampling and Sample Treatment: Ovine liver samples were purchased from local markets in Tarragona, Spain, and were chopped and blended with a domestic food blender. Homogenized samples were stored at -20 ºC prior to use. A scheme highlighting the entire sample treatment and analysis of the liver samples can be seen in Figure 1. Samples were defrosted overnight at 4 ºC and a 1 g aliquot was transferred into a 25-mL polypropylene centrifuge tube. The blank samples were spiked when appropriate by adding the working solution directly along with 10 mL of methanol. The resulting solution was vortexed for 5 min using a vortex mixer (Velp Scientifica) and then centrifuged at 1500 rpm for 10 min using a Genevac miVac Duo Concentrator. The supernatant was filtered using a 0.22-μm PTFE micro filter (Scharlab). The resulting solution was made up to 10 mL again with methanol and then diluted with water with 0.1% HAc to 50 mL. It was then frozen at -20 ºC overnight to promote precipitation of fats, lipids, and proteins. The solution was then centrifuged for 25 min and filtered under gravity to obtain a clear solution to submit to the mixed-mode SCX polymeric SPE cartridge for clean-up and concentration of the sample. The SPE protocol was as follows: the cartridge was conditioned with 5 mL of methanol and then by 2 × 5 mL portions of ultrapure water. The 50 mL sample solution was loaded and eluted with 10 mL of 4.5% NH4OH in methanol. The solution was directly injected into the LC–DAD system. During method validation, the extract was evaporated to dryness using a Genevac miVac Duo Concentrator, and reconstituted to 1 mL of mobile phase.


LC–DAD Conditions: Chromatographic analysis was performed using an Agilent 1100 HPLC system (Agilent Technologies) equipped with a diode array detector (DAD), a binary pump, and a 5 μL loop injector. Chromatographic separation was performed using a 150 × 0.46 mm, 5-μm Mediterranea Sea18 column (Technokroma). The mobile phase was as follows: water with 0.1% HAc ~ pH 2.8 (A) and acetonitrile (B). The flow rate was 1 mL/min, the monitoring wavelength was 270 nm, and the column thermostat was set at 30 ºC. The following gradient programme was used: 0–9 min from 10% to 55% (B); 9–11 min from 55% to 100% (B); 11–15 min continued at 100% (B) and then returned to initial conditions within 5 min.
Results and Discussion
Chromatographic Separation: Liquid chromatography (LC) coupled to a DAD detector was used for chromatographic separation, identification of analytes, and quantification. A DAD detector can be used initially because, although it is not as sensitive as MS, it is cheaper and more readily available, and if necessary, the developed method can be easily transferred to LC–MS because of the compatibility of the mobile phase. In addition, sulphonamides absorb light in the UV–vis region and the absorbance of the sulphonamides was evaluated from 220–360 nm. The monitoring wavelength was chosen to be 270 nm because this gave the strongest absorption for all sulphonamides evaluated (13,14). The mobile phase chosen was water with 0.1% HAc (A) and acetonitrile (B), which gave adequate separation. To obtain sufficient retention and separation, different elution gradients were tested and the gradient that gave the best separation of compounds is described in the experimental section. With this gradient all compounds were eluted in 8 min.
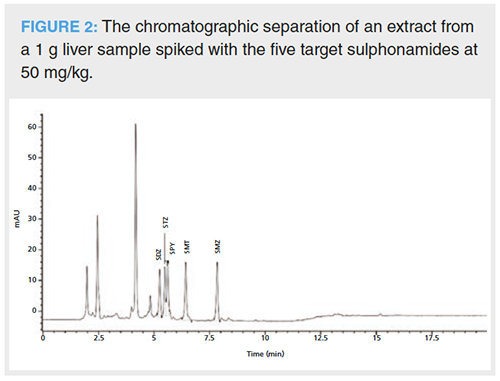
When applied to liver samples, a high water content at the beginning of the chromatographic run is needed to promote the elution of interfering hydrophilic substances, co-extracted from the complex liver matrix, in the first couple of minutes. This avoids coelution with analytes and limits noise. Figure 2 shows a chromatogram where an extract from a liver sample, spiked at 50 mg/kg with the analyte mixture, was injected. The elution of various interferences can be seen in the first 5 min of the chromatographic run. From 11–20 min after the gradient had ended, the chromatographic run was continued at 100% acetonitrile to clean the column and avoid a carry-over phenomenon in the following chromatographic runs (15). While this gradient provided the best possible separation, STZ and SPY-the second and third eluted compounds-overlapped slightly. However, resolution was calculated to be 1.87 ± 0.03 indicating they are sufficiently separated for this gradient programme to be used.

Extraction Method: Animal tissues, particularly the liver, are complex matrices that contain an abundance of interfering components such as fats, proteins, and carbohydrates. However, the liver is the site of detoxification and metabolism of sulphonamides, which give a good indication of residue levels of sulphonamides (16). To determine sulphonamides at low concentrations, effective clean-up steps can be utilized rather than increasing sensitivity through the use of expensive instrumentation. SPE is a well-developed procedure used for clean-up and concentration and is the most commonly used technique for food and environmental sample preparation to determine veterinary residues (17). Clean-up steps are essential in all samples and must be optimized before the extraction technique.
Solid-Phase Extraction: Two different SPE cartridges were evaluated for this procedure to concentrate the samples and clean up the complex liver matrix: a hydrophilic-lipophilic balanced copolymer SPE cartridge (150 mg), and a mixed-mode SCX polymeric SPE cartridge (150 mg). They were evaluated using initial conditions of 10 mL of aqueous solution spiked at 15 mg/L and eluting with 10 mL methanol. All these cartridges have been previously used to retain sulphonamides so that effective sample clean-up could occur; the hydrophilic-lipophilic balanced copolymer SPE cartridge is the most popular for food and environmental sample preparation for veterinary residues (17–20).
Under the initial conditions, both cartridges gave full recoveries of 92–130% for the mixed-mode SCX polymeric SPE cartridge and 109–125% for the hydrophilic-lipophilic balanced copolymer SPE cartridge. Previously, a silica-based modified with C18 SPE cartridge was also evaluated, which provided lower recoveries (35–90%) and had very poor reproducibility with relative standard deviations (RSD) of between 15–30%. Further optimization of both cartridges was achieved by increasing the sample volume and by decreasing the elution volume to concentrate the sample. For both cartridges, 50 mL was chosen as the optimal sample volume as increasing the sample volume up to this point did not affect recoveries. Larger volumes than 50 mL were not evaluated since the loading volume of SPE came from the extract for SLE. Moreover, 10 mL was chosen as the elution volume because decreasing it from 10 mL to 5 mL resulted in some compounds not completely eluting, in particular SDZ whose recoveries decreased by 37% in the mixed-mode SCX polymeric SPE cartridge and 25% in the hydrophilic-lipophilic balanced copolymer SPE cartridge. Two different organic elution solvents were tested, acetonitrile and methanol, and in the case of the mixed-mode SCX polymeric SPE cartridge, NH4OH was added to displace ionic interactions. In the mixed-mode SCX polymeric SPE cartridge both organic solvents gave similar recoveries, but the hydrophilicâlipophilic balanced copolymer SPE cartridge, methanol gave increased (11–13%) recoveries for each sulphonamide and was selected as the extraction solvent in both cases.
As the loading solvent in SPE is the extracting solvent in the previous SLE step, solvents other than water were also tested using standard solutions spiked at 15 mg/L for both SPE methods. The mixed-mode SCX polymeric SPE cartridge is a mixed mode sorbent functionalized with sulfonic groups so that it can establish cationic interactions with the target analytes. Therefore, organic solvent can be tested as the loading solvent. In contrast for the hydrophilic-lipophilic balanced copolymer SPE cartridge, dilution of the organic solvent with water or an evaporation step must occur before SPE. For this cartridge, dilution of methanol by a factor of four was found to significantly decrease recoveries, particularly for the most hydrophilic compounds as shown in Table 1. Therefore, an evaporation step is needed after SLE to completely remove the organic solvent and reconstitute the sample in water to be loaded into the hydrophilic-lipophilic balanced copolymer SPE cartridge.
For the mixed-mode SCX polymeric SPE cartridge, organic solvents methanol, EtAc, methanol with 0.1% HAc, and methanol–water with 0.1 HAc (25:75) were tested to load analytes onto the cartridge. Table 1 highlights that EtAc, methanol, and methanol with 0.1% HAc gave poor recoveries for SDZ, SMT, and SMZ. Although water with 0.1% HAc gave the highest percentage recoveries, 25:75 methanol–water with 0.1% HAc was chosen as the loading solvent because it gave similarly good recoveries, bar slightly lower for SDZ, and disregards the need for an evaporation step of the extract from SLE.
The recoveries and standard error for the two optimized SPE methods when applied to a 1 g liver sample spiked at 150 mg/kg, including the SLE extraction step of 10 mL methanol and 5 min extraction time, can be seen in Figure 3. For the hydrophilic-lipophilic balanced copolymer SPE cartridge, the resulting solution from SLE was evaporated to dryness and reconstituted in 50 mL of water. SPE clean-up was performed and compounds were eluted with 10 mL of methanol. For the mixed-mode SCX polymeric SPE cartridge, the resulting solution from SLE was diluted to 50 mL with water with 0.1% HAc (final solution was 25:75 methanol–water v/v) before SPE clean-up where compounds were eluted with 10 mL of methanol with 4.5% NH4OH. The mixed-mode SCX polymeric SPE cartridge was chosen because recoveries were higher with the exception of SDZ. In addition, this procedure is less time consuming than that of the hydrophilic-lipophilic balanced copolymer SPE cartridge due to the absence of an evaporation step.

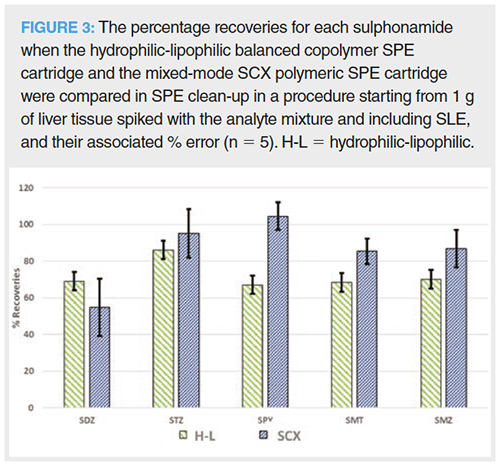
Solid-Liquid Extraction: Although PLE can be used to extract analytes from the solid matrix, it is expensive and unavailable in routine laboratories. Therefore, SLE with UAE and manual extraction were tested for the extraction of compounds from the liver. As SPE conditions were optimized before SLE conditions, methanol was chosen as the extraction solvent because it provided the best recoveries in SPE; no other organic solvents were tested. Thus, in both cases, 10 mL of methanol was added to 1 g of liver and the resulting mixture was shaken or sonicated for 5 min. The resulting solution was subjected to the optimized SPE clean-up procedure as the sample is too complex to be analyzed at this stage. The recoveries obtained are in general those depicted in Figure 3, with values ranging from 82% to 105% for all the compounds, with the exception of SDZ (56%).
Increasing the extraction time for longer than 5 min was found to have no effect on recoveries. Decreasing methanol volume from 10 mL to 5 mL resulted in lower recoveries (from 97% to 49%); therefore, methanol volume was kept at 10 mL.
Recoveries obtained for UAE and manual extraction aided with vortex were similar, between 61–106%. As a result, manual extraction was chosen as a more readily available method as UAE equipment was deemed unnecessary.
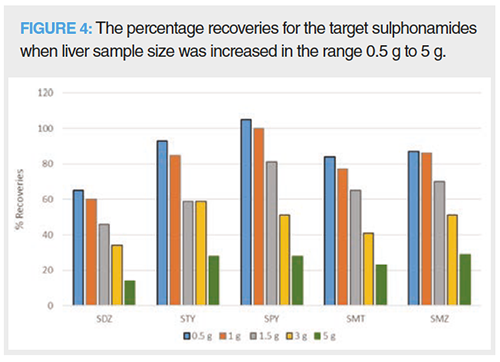
To lower the limits of detection (LOD), sample size can be increased. However, increasing sample size also increases the amount of interfering matrix components. Liver sample size was increased from 0.5 g to 5 g to test the optimal amount. A 1 g sample of liver gave good recoveries (60–100%); however, when the liver amount was increased to 1.5 g, the recoveries were lower (49–97%) as seen in Figure 4. Therefore, 1 g of sample was selected.

After SLE extraction with methanol, the sample was diluted with water prior to SPE extraction. However, this promoted the precipitation of fats and proteins. “Freezingâout” of interfering components was a simple and cheap method utilized by freezing extracts overnight and centrifuging for 25 min to remove interfering fats and proteins and provide a cleaner extract. Although this step is long, it allows many samples to be treated simultaneously.
The final optimized method consisted of taking 1 g of liver, adding 10 mL methanol, and vortexing for 5 min. The solution was then centrifuged and the supernatant was filtered with a 0.22-μm PTFE filter and made up to 10 mL again with methanol before diluting to 50 mL with water with 0.1% HAc. This solution was frozen overnight, centrifuged for 25 min, and then filtered under gravity to obtain a clear solution percolated in the mixed-mode SCX polymeric SPE cartridge. The SPE protocol was as follows: the cartridge was conditioned with 5 mL of methanol and then by 2 × 5 mL portions of ultrapure water. The 50-mL sample solution was loaded and eluted with 10 mL of 4.5% NH4OH in methanol. The solution was directly injected into the LC–DAD system. To further decrease the limits of detection, an evaporation step can occur after elution. This strategy was only used in the method validation. However, during the method optimization, samples were directly injected from the eluted 10 mL of 4.5% NH4OH in methanol because this is quicker and more straightforward than when it needed to be evaporated.
Method Validation: The final method was validated to demonstrate its performance by assessing linearity, percentage recoveries, repeatability, reproducibility, LODs, and limits of quantification (LOQ).
Instrumental calibration curves were first constructed from standard solution and good linearity from 0.05 or 0.1 mg/L to 30 mg/L was found for all analytes, with the value for the coefficient of determination (R2) being above 0.996 in all cases.
The final percentage recoveries were calculated by spiking the 1 g liver samples at 1 mg/kg and using the optimized procedure. Table 2 shows recovery values ranging from 78% to 99% with the exception of SDZ (64%). All RSDs were below 10% showing highly reproducible results. It should be mentioned that a non-spiked liver sample was first analyzed, and peaks at the same retention time to the target analytes were subtracted.
Method limit of quantification (MQLs) were calculated by applying the percentage recoveries as well as the concentration factor to the lowest point on the calibration curve. Method limit of detection (MDLs) were calculated by dividing this value by three and then subsequently spiking samples to obtain this final concentration to show that it is identifiable. Signal-to-noise ratio (S/N) criterion higher than three and 10 was used for MDLs and MQLs, respectively. All these results are displayed in Table 2.
The repeatability and reproducibility of the method were evaluated using five replicate extractions of 1 g of liver sample spiked at 2×MQL and performed on the same day or on different days, respectively. Both were expressed in terms of %RSD, which was 8–18% for repeatability and 9–29% for reproducibility.
As the MDL values obtained were 0.01–0.05 mg/kg, which is below the total MRL of 0.1 mg/kg for all sulphonamides issued by the EU Commission, this method can be used to confirm EU regulations are conformed to and it can also easily be transferred to LC–MS to lower MDLs.
Conclusions
Although a more sensitive technique, such as tandem mass spectrometry (MS/MS), can be used to obtain lower MDLs, this technology is not always available in routine laboratories. With respect to sample extraction, in this case it was found that cost could be cut by using more basic techniques, such as UAE and conventional SLE methods, which gave good recoveries, rather than methods using more expensive instrumentation, such as PLE and MAE.
It is evident that the complexity of the animal matrix is still creating a huge problem to produce effective methods to routinely comply with EU regulations. Finding effective sample clean-up and extraction techniques to reduce interfering matrix components is still an area open for improvement and exploration.
Acknowledgements
The authors would like to thank the Ministerio de Economía y Competitividad and the European Regional Development Fund (ERDF) (Project: CTQ2017-84373-R and CTQ2017-88548-P) for the financial support given.
References
- J. Aceña, S. Stampachiacchiere, S. Pérez, and D. Barceló, Anal. Bioanal. Chem.407, 6289–6299 (2015).
- L. Ramos, J. Chromatogr. A1221, 84–98 (2012).
- M. Núñez, F. Borrull, N. Fontanals, and E. Pocurull, Trends Anal. Chem.97, 136–145 (2017).
- M. Pérez-Rodríguez, R. Gereado Pellerano, L. Pezza, and H. Redigolo-Pezza, Talanta182, 1–18 (2018).
- A. Larivière, S. Lissalde, M. Soubrand, and M. Casellas-Français, Anal. Chem. 89, 453–465 (2017).
- P. Vázquez-Roig and Y. Picó, Trends Anal. Chem.71, 55–64 (2015).
- B.K. Tiwari, Trends Anal. Chem.71, 100–109 (2015).
- A.G. Frenich, M.D. Aguilera-Luiz, J.L.M. Vidal, and R. Romero-González, Anal. Chim. Acta661, 150−160 (2010).
- M. Solliec, A. Roy-Lachapelle, and S. Sauvé, Anal. Chim. Acta 883, 415–424 (2015).
- E. Miró-Abella, P. Herrero, N. Canela, L. Arola, M.R. Ras, F. Borrull, and N. Fontanals, J. Chromatogr. B1105, 47–53 (2019).
- A. Freitas, J. Barbosa, and F. Ramos, J. Chromatogr. B976, 49–54 (2015).
- European Commission, Off. J. Eur. Union L15, 1–72 (2010).
- Q. Zou, M. Xie, Y. Liu, J. Wang, J. Song, H. Gao, and J. Han, J. Sep. Sci.30, 2647–2655 (2007).
- K. Deng, X. Lan, G. Sun L. Ji, and X. Zheng, Food Anal. Methods9, 3337–3344 (2016).
- M.T. Martins, J. Me, F. Barreto, R.B. Hoff, L. Jank, M.S. Bittencourt, J.B. Arsand, and E.E.S. Schapoval, Talanta129, 374–383 (2014).
- H. Abdallah, C. Arnaudguilhem, F. Jaber, and R. Lobinski, J. Chromatogr. A1355, 61–72 (2014).
- A. Andrade-Eiroa, M. Canle, V. Leroy-Cancellieri, and V. Cerdà, Trends Anal. Chem. 80, 641–654 (2016).
- M. Periša and S. BarbiÄ, J. Sep. Sci.37, 1289–1296 (2014).
- H. Sun, L. Ai, and F. Wang, Chromatographia66, 333–337 (2007).
- W. Hela, M. Brandtner, R. Widek, and R. Schuh, Food Chem.83, 601–608 (2003).
Leah Walker is currently completing her MChem degree at the University of Edinburgh.
Francesc Borrull is Full Professor in analytical chemistry in the Analytical and Organic Chemistry Department at the Rovira i Virgili University in Tarragona since 2003.
Eva Pocurull is Assistant Lecturer in analytical chemistry in the Analytical and Organic Chemistry Department at the Rovira i Virgili University in Tarragona since 2000.
Núria Fontanals is Senior Researcher in the Analytical and Organic Chemistry Department at the Rovira i Virgili University in Tarragona since 2007.









Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

