Robustness Modelling in Ultrahigh-Pressure Liquid Chromatography Methods
The Column
Many workers in pharmaceutical laboratories are unable to change any aspect of their methods, although they often encounter severe problems and create many out-of-specification (OoS) results. They are particularly afraid to investigate these problems from a chromatographic perspective in case they generate new unforeseen problems. In the literature, however, there are numerous examples showing that it is worthwhile trying to understand the reasons for “unexplainable” behaviour in ultrahigh-pressure liquid chromatography (UHPLC) using modelling. By using modelling, problems can be recognized and often eliminated with legal operations according to the allowed tolerance limits mentioned in pharmacopoeia descriptions. The following article aims to show that “visual chromatographic modelling” can be a useful aid.


Photo Credit: Elesey/Shutterstock.com

Many workers in pharmaceutical laboratories are unable to change any aspect of their methods, although they often encounter severe problems and create many out-of-specification (OoS) results. They are particularly afraid to investigate these problems from a chromatographic perspective in case they generate new unforeseen problems. In the literature, however, there are numerous examples showing that it is worthwhile trying to understand the reasons for “unexplainable” behaviour in ultrahigh-pressure liquid chromatography (UHPLC) using modelling. By using modelling, problems can be recognized and often eliminated with legal operations according to the allowed tolerance limits mentioned in pharmacopoeia descriptions. The following article aims to show that “visual chromatographic modelling” can be a useful aid.
A. Schmidt et al. (1) discussed the drug pramipexol, which was showing repeated out-of-specification (OoS) results. Sometimes it was a turnover in peak elution order of two neighbouring peaks, sometimes they coeluted. The reason for this observation was that the set point (SP) was close to the edge of failure (EoF) at an extreme part of the design space (DS). This unstable condition could be found with the help of modelling software and corrected by reducing the concentration of the ion pair reagent to prevent the coelution and peak turnover happening again.
Another common problem is the fear of changing a method, even when it is obvious that the method is out of date, takes too long, and exhibits often unexplainable OoS findings, resulting in weeks of lost time and a lot of confusion (1,2). With modelling these problems can often be resolved in a fast and efficient way (3).
What Can We Change in a Validated Method?
What can be legally changed in methods can be found in the United States Pharmacopeia (USP) and in the European Pharmacopoeia (Ph. Eur.). Chapter 2.2.46 of Ph. Eur. “Chromatographic Separation Methods” contains the following description: “These allowed adjustments may be necessary because the stationary phases are described in a general way, and there are a variety of phases available commercially that meet these general descriptions, which can result in chromatographic behaviour differences” (4). Chapter 2.2.46 is similar to the USP Chapter 621, concerning the adjustments allowed.
Many workers are concerned that quality by design (QbD) compatible modelling in the analytics means that criticism of QbD in the production would also follow, but ICH Q8R2 states explicitly that more knowledge based on solid science causes less regulatory oversight (Figure 1).


UHPLC Method Adjustments
Heiko Behr wrote in his blog: “The last liquid chromatography allowed adjustments revision in 2010, stated that adjustments for gradient methods are more critical than isocratic methods. These changes can lead to shifts in peaks and to a different step of the gradient. This then leads to the incorrect assignment of peaks, peak masking, or an elution shift that occurs beyond the prescribed elution time. As a result, the allowed adjustments were classâdivided for isocratic and gradient methods, with minor allowed adjustments for the latter. Effective from August 2014 (USP37-NF32, 1st supplement) the USP split the allowed adjustments into isocratic and gradient sections. In addition, the USP introduced a substantial change in the column related to allowable adjustments for isocratic methods to improve user flexibility” (4).
The primary focus is keeping the column plate number, and thus resolution, fairly constant. Similarly, to the last revision of the USP 621 chapter, the L/dp ratio was introduced for maintaining nearly constant efficiency and, therefore, resolution. But this change is not only valid for isocratic elution (like in the USP), it is also customized to gradient methods. This explanation is very much valid in gas chromatography (GC). In reversed phase chromatography, the selectivity is governed by interactive forces in the aqueous eluent (6).
Behr is showing the differences between the allowed adjustments for isocratic and gradient liquid chromatography methods for the new Ph. Eur. Draft, the current Ph. Eur. Supplement 9, and the current USP 40-NF35 in useful tables (4). The “allowable changes” are sometimes vague and would lead in several cases to OoS results. The only way to make changes should be done by modelling based on a few basic experiments around the SP, which would discover how peak movements occur and from there one can return in modelling to the original validated system suitability test (SST), which is validated. If the adjustment is not possible because the deviation is too large, then a revalidation has to be performed. This is currently not a problem because the 50–100-mmâlong columns of small particle sizes (dp: 1.7–1.8 µm) have analysis times typically below 10–15 min, so the validation would not require more than a few days. It is also important that the model is included in the new master file, allowing later adjustments because alterations of the SP inside of the DS are not considered to be a “change”, but an adjustment to meet system suitability parameters.
UHPLC Method Robustness
The communication with regulatory bodies includes relatively high handling charges for “post approval changes” and are connected with a great deal of bureaucracy. Therefore, pharmaceutical and food companies are working to improve the robustness of their methods, so they can safely be used in routine work. Nevertheless, methods developed by trial and error are always subject to many OoS events, causing a breakdown in production and interrupting the provision of new, promising drugs to patients.
Regulatory agencies are therefore required to prove the robustness of new UHPLC methods. Earlier it was sufficient to submit methods with decent statistics, today the regulatory agencies want to know how the method was developed and why the pH, gradient shape, temperature, or the flow rate were selected and localized specifically at a given SP in the submissions.
In 2006 the regulatory expectations changed to more scientific fundamentals. The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) mentions the regulatory expectations in the ICH Q8, R2 towards more scientifically reliable drug master files. QbD should now be introduced in the analytical work.


What is QbD?
QbD is the end of trial and error. The UHPLC method development process with QbD principles should be properly documented and the method should be based on solid science. The work should be carefully planned, instead of based on the “intuitions” of “experienced” users in the laboratory. This includes registering all starting factors, such as the batch number of the stationary phase, the serial number of the column, the instrument configuration, type of pumps, injection devices, and detectors, all in one data set, including parameter tolerances. These are included in the installation qualification (IQ) and should be regularly revisited. Another problem is that laboratory workers often forget some details and therefore have to repeat the experiments several times. The best way to get a precise description of the method conditions is to collect them from the instrument in a method report.
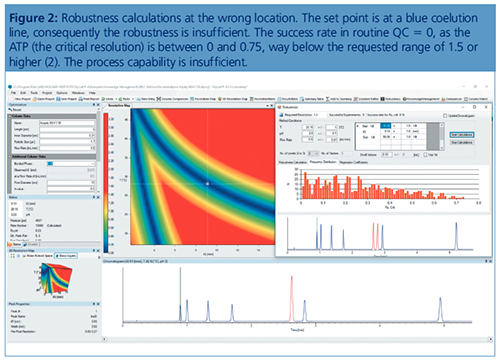
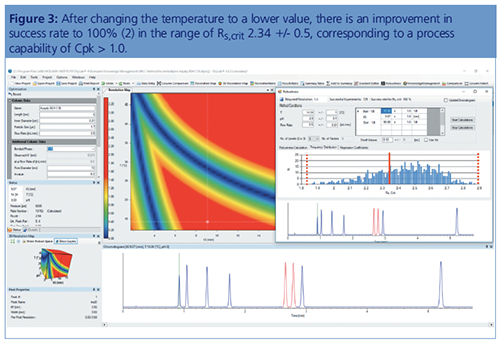
All this was leading to an automated generation according to different design of experiments (DoEs), which was started by modelling tools, where the UHPLC instrument was preprogrammed to perform a number of runs in a sequence, followed by an evaluation of the results. The question is, however, how to get as close as possible to the separation optimum? Even in the case of a sufficient separation, variabilities of the parameter could interact with each other and the analytical target profile (ATP), often the critical resolution, would fall below the validated level of 1.5 (baseline separation).
Currently, a submitted method has to be validated, which means the applicant has to approve that the method is well suited for its intended use. However, many times in the validation process, which consists of a large number of runs for statistical purposes, one obtains OoS results. One realizes, often too late, that the method was not robust at all and the method has to be redeveloped. Therefore, we should now include the robustness tests before the validation step. Modelling software can test all the eventual problems in a much shorter time than one would be able to do in the laboratory.
What is More Important, Knowledge of Statistics or Knowledge of UHPLC?
The experienced user says that if the chromatographic method is robust, one would never have any problems with the statistical evaluation of the method in the production process.
How is it Possible to Make a New Submission According to the Above Principles?
In the ICH Q8, R2 there is a request to use solid science in the submissions. Therefore, a scientific model of the chromatographic history of the method should be included in the submission in a “Knowledge Management Document”, including the experiments for the model, which would allow later potential changes in the SP. As far as the SP remains inside of the DS, no revalidation will be necessary. This will allow production costs to be reduced and make method handling more flexible. The cooperation with regulatory agencies will become more relaxed and harmonious.
Acknowledgement
The authors are thankful to Lloyd R. Snyder for his fundamental contributions to DryLab, and to Waters and Shimadzu for their support of this work.
References
- Alexander H. Schmidt, Mijo Stanic, and Imre Molnár, Journal of Pharmaceutical and Biomedical Analysis91, 97–107 (2014).
- Róbert Kormány, Imre Molnár, and HansâJürgen Rieger, Journal of Pharmaceutical and Biomedical Analysis80, 79–88 (2013).
- Alexander Schmidt and Imre Molnár, Journal of Pharmaceutical and Biomedical Analysis78–79, 65–74 (2013).
- Heiko Behr, https://phenomenex.blog/2017/09/13/european-pharmacopeia/Phenomenex, Revision of European Pharmacopeia (EP) Chapter 2.2.46.
- A. Hussain, “Pharmaceutical Quality in the 21st Century: Personal Reflections,” https://www.aegiscorp.com/files/presentations/
- Csaba Horváth, Wayne Melander, and Imre Molnár, Journal of Chromatography125, 129–156 (1976).
Imre Molnár is founder and president of Molnár-Institute for Applied Chromatography. Having received a doctorate degree from German Saarland University in analytical chemistry, Dr. Molnár joined Csaba Horváth’s research team at Yale, resulting in numerous publications such as the “Theory of Solvophobic Interactions” and on the fundamentals of reversed phase chromatography. After returning to Europe he founded the Institute for Applied Chromatography in Berlin in 1981. Since 1984, joining with Lloyd Snyder and John Dolan, he has focused on in silico method development in HPLC, resulting in the software DryLab. Since then, more than 200 peerâreviewed papers on DryLab’s scientific and industrial application have been published.
Alexander H. Schmidt is General Manager and Director of the Quality Unit at Chromicent GmbH in Berlin. He offers a diverse array of experience in all stages of the life cycle of analytical methods: from method development in a quality by design framework, to routine use of the method in analytical instrument qualification and GMP compliance, and maximizing productivity in the laboratory. Over the years, he has published numerous articles on HPLC and UHPLC method development for pharmaceuticals and complex natural compound mixtures. He is also a guest lecturer at the Molnar Institute of Applied Sciences.
E-mail: info@molnar-institute.comWebsite:www.molnar-institute.com














Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

