Quantitative Proteomic Workflow for Discovery of Early Rejection Kidney Transplant Peptide Biomarkers and Subsequent Development of SRM Assays in Urine
Special Issues
The accurate diagnosis of renal allograft rejection currently depends upon a biopsy. Transplant medicine would benefit greatly from the availability of noninvasive tests for early detection of rejection and immunosuppressive drug therapeutic monitoring. Only a limited number of studies have been published to date on specific proteins associated with allograft rejection. Typically, renal dysfunction due to humoral transplant rejection or other pathologies results in the increase of protein excreted in urine (1–5). In blood, endogenous peptides (not generated by trypsin digestion ex vivo) are likely candidate biomarkers for many diseases and pathologies as they are secreted from tissues and enter the bloodstream (6,7). The analysis of endogenous protein and peptide fragments in urine can provide a noninvasive, early indication of kidney transplant rejection or disease.
Endogenous peptide recovery from body fluids poses numerous hurdles. First, proper collection and storage must minimize the ex vivo generation of artifactual peptides. Second, the dynamic range of molecular size and abundance requires separation of proteins from peptides and metabolites.
Recently, there has been an increased focus in the clinical community on the development of targeted selected reaction monitoring (SRM)–based assays as an alternative to traditional but less specific enzyme-linked immunosorbent assay (ELISA). Mass spectrometry (MS)–based assays offer a number of alternatives over antibody-based assays, including higher specificity, robust quantification based upon heavy-labeled standards, and the ability to monitor a panel of diagnostic markers in a multiplexed assay format. Unfortunately, designing effective targeted assays remains a challenge, and can require large numbers of samples and multiple iterations to empirically determine the optimal proteotypic peptides and transitions. Fortunately, in many cases, SRM experiments are preceded by discovery-based experiments to develop a list of target proteins and peptide biomarkers. High-resolution accurate mass MS-MS spectra generated in discovery experiments can be used directly to increase the efficiency and accuracy of SRM assay development by guiding the selection of optimal transitions (Figure 1).


Figure 1: Workflow for integrating differential expression analysis with targeted protein quantitation.
Goal
This study describes the development of a workflow utilizing off-line size fractionation coupled with on-line liquid chromatography and high-resolution tandem MS on a hybrid mass spectrometer specifically geared toward the identification of differentially expressed proteins and endogenous peptides in urine. Selected protein or peptide biomarkers identified in the high-resolution discovery workflow were subsequently used to develop targeted SRM assays on a triple-quadrupole mass spectrometer.
The described quantitative workflows were used to analyze urine samples from normal, early rejection, and acute humoral rejection transplant patients.
Experimental Conditions and Methods
Sample Preparation
Urine samples were collected from study participants with full consent and approval under IRB protocols. The samples were transported frozen and thawed before sample processing. Urine samples were concentrated using 2-mL single-well devices (Vivaspin 2 3 kDa MWCO, Vivascience, Massachusetts). Concentrated urine samples were diluted 4:1 with 8 M guanidine HCl, 10 mM dithiothreitol (DTT), 150 mM tris HCl (pH 8.5, adjusted at 20 °C) and mixed before incubation at 37 °C for 1 h. The denatured samples were then alkylated with 45 mM iodoacetic acid (500 mM stock concentration in 1 M ammonium bicarbonate) in the dark for 1 h at room temperature. The residual alkylation agent was then reacted with 15 mM DTT followed by adding trifluoroacetic acid to a final concentration of 2% (v/v) to acidify the sample.
Sample Desalting Employing C18 Reversed-Phase Resin
HyperSep-96 C18 solid-phase extraction (SPE) media (Thermo Fisher Scientific, Waltham, Massachusetts) was used to desalt the denatured, reduced, and alkylated samples prepared in the above step. A 25-mg SPE device was adapted for single use by centrifugation (500 g for 3–5 min) or in a 96-well format using a plate adaptor and a HyperSep universal vacuum manifold. In both cases the centrifugation and vacuum conditions were adjusted to achieve a 0.5–1 min residence time in the media bed. Long centrifugation times at high speeds or prolonged vacuum processing should be avoided to ensure that the reversed-phase resin does not dry out, requiring rewetting with organic solvent. The HyperSep C18 resin was conditioned before use with the following procedure:
1. Fill the SPE device with n-propanol and pass though C18 media slowly (1–2 min). Repeat two times.
2. Follow with 0.25% (v/v in water) trifluoroacetic acid to rinse the whole SPE device. Repeat two times. Load 100–125 μL of sample (equivalent to 20–25 μL of original serum sample) prepared as described earlier and process by centrifugation or vacuum to flow through the SPE media for a residence time of 0.5–1 min. Prepare a wash buffer of 8M guanidine HCl, 10 mM dithiothreitol (DTT), 150 mM tris HCl (pH 8.5, adjusted at 20 °C) diluted 4:1 with trifluoroacetic acid to a final concentration of 0.25% (v/v). Fill the SPE device and pass through the resin bed as described earlier. Note: This step removes the nonadsorbed sample in the interstitial fluid space within the SPE packed bed. Wash 3× with 0.25% (v/v) trifluoroacetic acid as described earlier.
Elute the desalted sample with 500 μL of 50% (v/v) n-propanol in 0.1% (v/v) formic acid by centrifugation (single devices) or vacuum filtration.
Sample Cleanup Using Ultrafiltration Membranes
Sample cleanup by ultrafiltration was carried out in single-well devices (Microcon 50-kDA MWCO membrane, Millipore Corporation, Billerica, Massachusetts) using a microcentrifuge. Multiple single-well ultrafiltration devices can also be loaded into a carrier plate (Millipore Corporation) and processed by vacuum. Multiwell plates fitted with a 30-kDA MWCO membrane (Vivascience MA, Sartorius Stedim Biotech SA) also were used with larger numbers of samples on a vacuum manifold. Before use, all ultrafiltration membranes and devices were washed with 50% (v/v) n-propanol, 0.1% (v/v) formic acid.
Mass Spectrometric Analysis
Samples in 5% (v/v) acetonitrile 0.1% (v/v) formic acid were injected with a Thermo Scientific Micro Autosampler onto a 25 cm × 75 μm fused-silica capillary column packed with Hypersil GOLD aQ 5 μm media, a Thermo Scientific Surveyor MS pump running with a flow splitter (1:1000), in a 250-μL/min gradient of 5% (v/v) acetonitrile, 0.1% (v/v) formic acid to 30% (v/v) acetonitrile, 0.1% (v/v) formic acid over the course of 180 min with a total run length of 240 min. An LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific) was run with a 60 K resolution full scan with 1e6 target values and 1000 ms inject times with up to five precursor ions selected for MS-MS. HCD MS-MS spectra at 15 K resolution were acquired with data-dependent 8e4 trigger thresholds, 2e5 target values, and 2000-ms injection times. A normalized collision energy of 45 was used. Collision-induced dissociation (CID) MS-MS spectra were acquired with 3e4 target values, 1e4 data-dependent trigger thresholds. In both experiments, monoisotopic precursor selection is enabled, with +1 and unassigned charge states rejected. Label-free differential analysis was performed using the SIEVE algorithm.
Development of SRM Assay
Methodology
The mass spectrometer was equipped with a Thermo Scientific Surveyor MS pump, and Micro Autosampler (flow split preautosampler as used in the discovery instrument format). The source was the IonMax source equipped with a low-flow metal needle, with a 50 mm × 1 mm Hypersil GOLD aQ column. Solvent A was Fisher Optima LC–MS grade water with 0.2% (v/v) formic acid, and solvent B was Fisher Optima LC–MS grade 30% (v/v) acetonitrile with 0.2% (v/v) formic acid. The peptides found to be differentially present in the discovery data were imported directly into SRM Workflow software. Transitions were based upon predominant fragments found in the discovery data (>five transitions per peptide), overlapped with the predominant ion fragments SRM builder. Alignment and relative quantification of the transitions were performed with SRM builder.
Results and Discussion
Analysis of High-Resolution MS-MS Data by SIEVE
Although lower-resolution trap-based CID scans can be triggered on lower-abundance peptides, when searching a database "no enzyme," the large number of potential misidentifications, even with high parent-mass accuracy, makes for a difficult high-confidence sequence assignment. This, coupled with high charges and uneven charge distributions (unlike the case when peptides digested with trypsin), makes for a difficult assignment and an even more difficult prediction of a useful SRM transition. Using the HCD (Higher-Energy Collisional Dissociation) cell to perform the fragmentation of these difficult to analyze peptides yields an accurate-mass product ion with an easily assignable charge state.


Figure 2: Reconstituted ion chromatograph data analysis of high-resolution MS-MS data by SIEVE: (a) Nondifferentially expressed putative biomarker (ratio 0.8, mitochondrial ribosomal protein). (b) Increased expression (ratio 167.4) of putative biomarker.
Typical reconstructed ion chromatograms from nondifferentially expressed (Figure 2a) and highly overexpressed (Figure 2b) putative protein markers are shown in Figure 2. A summary of the top 31 peptides/proteins increased in stable versus prerejection (putative early markers) and stable versus rejecting (putative late markers) are shown in Table I. The majority of peptides were up-regulated in the disease state. Few peptides were down-regulated or equivalent (ratio close to 1). This result is consistent with the hypothesis that endogenous peptide fragments represent a "metabolic snapshot" of proteolytic activities induced by disease or inflammation. Presumably, proteolytic fragments are excreted into the urine and can be a rich source of disease biomarkers. In this case, only an increase and not a decrease in endogenous peptides would be expected in disease samples.
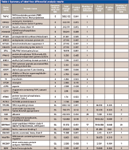
Table I: Summary of label-free differential analysis results
Development of a Discovery-Driven Selected-Reaction Monitoring (SRM) Assay on a Triple-Quadrupole Mass Spectrometer
High-resolution MS-MS spectra of target proteins and peptides illustrated in Figure 3a were analyzed to determine the most sensitive or intense transitions. The selected peptides and transitions were used to build an SRM method for a triple-quadrupole system with SRM Workflow software (Figure 3b). Individual transitions for a vitronectin peptide (Figure 3c) and a uromodulin peptide (Figure 3d) are illustrated. Preliminary results are summarized in Figure 4 for the putative biomarkers vitronectin and uromodulin in prerejection — and rejecting — patient samples. The five transitions for the uromodulin peptide, charge state +3, 590.007 (but not charge state +2, 884.507) were observed in the prerejection — and rejecting — patient samples but not in the stable patient samples. This demonstrates the presence of a specific endogenous peptide in a patient sample under early and full rejection conditions. The absence of the same endogenous peptide in the stable-patient samples suggests that it might be a useful marker for early rejection, although the sample set is very small and much further verification is needed. The confidence level for this result is high because all five transitions for the peptide were observed. The probability of this result due to random chance is very low.

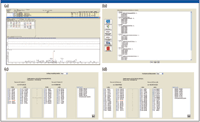
Figure 3: Discovery driven design of SRM assay method: (a) High-resolution MS-MS spectra of target proteins/peptides. (b) Building a method for the Thermo Scientific TSQ Quantum Ultra with SRM Builder software. (c) Transitions for a vitronectin peptide. (d) Transitions for a uromodulin peptide.
Conclusion
A robust workflow was created for the recovery of intact endogenous peptides (peptides not generated by proteolytic cleavage ex vivo) from urine. To examine the effectiveness of this workflow, quantitative, label-free differential analysis was applied to high-resolution MS spectra to drive the selection of putative peptide markers for early transplant rejection. Obtaining the high-resolution accurate-mass MS-MS fragmentation data facilitated design of a targeted SRM assay by yielding highly accurate fragment assignments on difficult-to-analyze endogenous peptides. A targeted SRM assay for one of the peptides in the panel was tested on the patient sample set and quantitative data were obtained, showing that the described workflows can serve as a model to link biomarker discovery and validation.

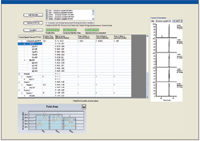
Figure 4: SRM assay results for uromodulin peptide transitions in prerejection and rejecting clinical samples.
David Sarracino, Bryan Krastins, Amol Prakash, and Mary F. Lopez are with Thermo Fisher Scientific BRIMS Center, Cambridge, Massachusetts.
Waichi Wong and Emmanuel Zorn are with Massachusetts General Hospital, Harvard, Boston, Massachusetts.
Michael Athanas is with Vast Scientific, Cambridge, Massachusetts.
References
(1) M.M. van Timmeren, V.S. Vaidya, R.M. van Ree, L.H. Oterdoom, A.P. de Vries, R.O. Gans, H. van Goor, C.A. Stegeman, J.V. Bonventre, and D.J. Bakker, Transplantation 84, 1625–1630 (2007).
(2) W.S. Oetting, T.B. Rogers, T.P. Krick, A.J. Matas, and H.N. Ibrahim, Am. J. Kidney Dis. 47, 898–904 (2006).
(3) J. Kaden, J. Groth, G. May, and B. Liedvogel, Urol. Res. 22, 131–136 (1994).
(4) A.M. Teppo, E. Honkanen, P. Finne, T. Tornroth, and C. Gronhagen-Riska, Transplantation 78, 719–724 (2004).
(5) G. Filler, F. Priem, N. Lepage, P. Sinha, I. Vollmer, H. Clark, E. Keely, M. Matzinger, A. Akbari, H. Allthaus, and K. Jung, Clin. Chem. 48, 729–736 (2002).
(6) M.F. Lopez, S. Kuzdzal, A. Mikulskis, E. Golenko, E.F. Petricoin III, L.A. Liotta, W.F. Patton, C. Lynch, R. Gordon, G.R. Whiteley, K. Rosenblatt, P. Gurnani, A. Nanda, S. Neill, S. Cullen, M. O'Gorman, D. Sarracino, and D. Fishman, Clin. Chem. 53, 1067–1074 (2007).
(7) M.S. Lowenthal, A.I. Mehta, K. Frogale, R.W. Bandle, R.P. Araujo, B.L. Hood, et al., Clin. Chem. 51, 1933–1945 (2005).
















Free Poster: NDSRI Risk Assessment and Trace-Level Analysis of N-Nitrosamines
April 25th 2025With increasing concern over genotoxic nitrosamine contaminants, regulatory bodies like the FDA and EMA have introduced strict guidelines following several high-profile drug recalls. This poster showcases a case study where LGC and Waters developed a UPLC/MS/MS method for quantifying trace levels of N-nitroso-sertraline in sertraline using Waters mass spectrometry and LGC reference standards.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.
Using the Carcinogenic Potency Categorisation Approach (CPCA) to Classify N-nitrosamine Impurities
April 25th 2025Learn how to manage nitrosamine impurities in pharmaceuticals with our free infographic. Discover how the CPCA approach establishes acceptable intake limits and guides the selection of NDSRI reference samples. Stay compliant and ensure safety with our ISO-accredited standards.

