Quantitating Toxaphene Parlar Congeners in Fish Using Large Volume Injection Isotope Dilution GC with Electron-Capture Negative Ion MS
LCGC North America
A novel automated ultratrace gas chromatography–mass spectrometry (GC–MS) method has been developed that quantitates the eight toxaphene Parlar congeners designated in U.S. Environmental Protection Agency Method 8276. This method, combined with an efficient extraction, cleanup, and fractionation technique, makes is possible to extend instrument detection limits to the low parts-per-trillion concentration level for these toxaphene Parlar congeners.
This article describes a novel automated ultratrace gas chromatography–mass spectrometry (GC–MS) method that quantitates the eight toxaphene Parlar congeners as designated in U.S. Environmental Protection Agency (EPA) Method 8276. This method was developed and applied to fish tissue and used a conventional sample preparation approach to extract, clean up, and fractionate selected Parlar congeners. Instrument detection limits for the eight toxaphene Parlar congeners quantitated were calculated in two ways. Quality control (QC) measures that were used to validate the new method are discussed and QC results are presented for these eight Parlar congeners of environmental significance.
Toxaphene was introduced as a pesticide in the United States in the 1940s and subsequently banned in 1990 and banned globally in 2001 by the Stockholm Convention on Persistent Organic Pollutants. Toxaphene usage was increased because of its substitution for dichlorodiphenyltrichloroethane (DDT) on cotton crops from 1972 to the early 1980s. Toxaphene consists of a complex mixture of polychlorinated monoterpenes (primarily bornanes and camphenes) produced commercially from 1947 to 1982 and purported to contain 600-plus separate congeners (1). "Weathered toxaphene" may only appear to be present because of degradation products such as Hex-SED and Hep-SED (refer to Table I for systematic nomenclature). Several of these numerous congeners that persist in the environment are currently assigned "Parlar" numbers (2). Quantitating specific Parlar congeners has been an active area of pursuit (3,4).



In our study, a gas chromatograph was interfaced to a single-quadrupole mass spectrometer that used electron-capture negative ionization (ECNI). Sample extracts were injected via a multipurpose single rail autosampler using a large volume, split vent–stopped flow injection technique (LVI-SVSF). Quantitative analysis was facilitated by using isotope dilution (ID). Some Parlar congeners, such as P62, are thermally degraded in hot inlets. LVI-SVSF enables a sample extract to be injected in a cooled inlet (40 °C) without thermally breaking down P62.
Experimental
Unlabeled stock solutions, purchased as certified reference standards (LGC Promochem and Cambridge Isotope Laboratories), were diluted in isooctane and stored in sealed vials at -20 °C until used as calibration standards and spikes. The 13C-labeled Parlar 26, 13C-labeled Parlar 50 (used as internal standards), and 13C-labeled Parlar 62 (used as a surrogate) were obtained from Cambridge Isotopes Laboratories. A continuing calibration verification (CCV) standard that contained all eight Parlar congeners was prepared. The concentration of each Parlar congener in the CCV was prepared to be at or near to the midpoint of the range of calibration standards.
Sample Preparation
All samples were frozen upon arrival at the laboratory. Acceptable samples included whole fish, skin-on edible fish fillets, and skin-off edible fish fillets. Fish samples were spiked with the surrogate and then homogenized and extracted following in-house methods. These sample preparation methods used a Dionex ASE 300 accelerated solvent extractor (Thermo Fisher) (5). Extracts were further processed through Florisil cleanup and fractionated using activated silica gel. This liquid–liquid extraction approach with extensive cleanup and fractionation was adapted and modified from an earlier published method (6). Four total fractions were prepared. Fractions 2 and 3 contained the Parlar congeners with a final extract volume of 2.0 mL, respectively. Fraction 2 contained predominantly P26 while fraction 3 contained all of the other congeners. Fractions 2 and 3 were concentrated down to ~0.5 mL, solvent exchanged to isooctane, and then adjusted to a final extract volume of 2.0 mL. Next, 20 μL of fraction 2 and 20 μL of fraction 3 were added into a clean Waters Total Recovery vial that contained 10 μL of internal standard. This procedure gave a final volume of 50 μL. Sample extracts were stored at refrigerated temperatures (~4 °C) before analysis.
Instrumentation
A 6890/5973N inert GC-MSD (Agilent Technologies) gas chromatography–mass spectrometry (GC–MS) system that incorporates an LVI-SFSV inlet with electronic pressure control was used. The system's mass selective detector was converted to ECNI-MS by the installation of a chemical ionization ion source (Agilent Technologies). A tank of high-purity pressurized methane gas was attached to the reagent gas fitting in back of the detector. Methane was introduced at a gas pressure of ~10 psi. The detector was operated under negative ion conditions to generate the quantitative data. The GC–MS system and CIS-4 inlet (Gerstel, GmbH & Co. KG) were interfaced to a personal computer (PC). ChemStation (Agilent Technologies) software for chromatographic control, data acquisition, and storage was factory installed on the PC. Chromatographic separation of Parlar congeners was achieved using a 30 m x 0.25 mm, 0.25-μm df DB-XLB (Agilent Technologies) open tubular column and a multistep oven temperature program ramp as detailed below (7). The GC–MS conditions were as follows:
- GC oven temperature program: 60 °C (hold for 1 min) to 200 °C at 10 °C/min, then to 230 °C at 1.5 °C/min, then to 300 °C at 10 °C/min, then to 340 °C at 45 °C/min (hold for up to 10 min). The total run time was 53 min.
- Ion source temperature: 150 °C; transfer line temperature: 230 °C
- Selective ion monitoring (SIM) was used under the timed group option
- Optimal EM volts for ultrahigh sensitivity: ~2200 V; trace ion detection: on; resolution: low
- Methane (CH4) was used as a reagent gas at ~10–15 psi. The Agilent 5973N system was operated in negative ion mode.
- In contrast to electron ionization mass spectrometry (EI-MS), chemical ionization mass spectrometry (CI-MS) does not require frequent tuning. The Agilent 5973N mass spectrometer, operated in the CI-MS mode, was tuned when it seemed to perform poorly. Tuning (using the process described in the instrument's manual) approximately once per month is appropriate.
A dual syringe robotic autosampler system (MPS-2, Gerstel, GmbH & Co. KG) was mounted on top of the GC–MS system. Maestro (Gerstel, GmbH & Co. KG) software that controls operation of the autosampler was integrated into the ChemStation software.
Summary of LVI-SVSF
First, 10 μL liquid extract (sample) was injected into a cooled inlet (40 °C) at zero inlet pressure while keeping the split vent open (solvent vent at 100 mL/min). At 0.5 min after injection, the split valve was closed. The inlet was rapidly heated to 275 °C under splitless conditions. The split vent was opened 2.5 min after sample injection with a purge flow to split vent of 50 mL/min. This technique enables sample volumes greater than 1 μL to be injected without chromatographic peak distortion while increasing analyte sensitivity 10-fold.
Results and Discussion
Method Development
This method improves on the United States Environmental Protection Agency's (U.S. EPA) Method 8276 in three ways:
- LVI replaces the conventional 1–μL injection into a hot inlet;
- 13C isotopically labeled Parlar congeners P26 and P50 replace the single PCB 204 congener as internal standards; and
- 13C P62 is added as a surrogate instead of using decachlorobiphenyl to continuously monitor analyte recovery.

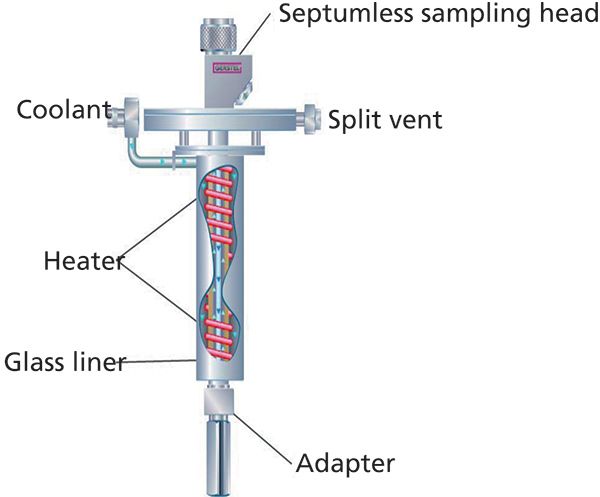
Figure 1: Schematic diagram of the Gerstel CIS-4 cooled inlet septumless injector. (Courtesy of Gerstel, Inc.)
The principles and advantages of LVI-GC versus conventional 1-μL hot injection are well understood (8). The application of a cooled septumless inlet coupled to the SVSF technique while incorporating an open tubular column interfaced to a highly selective and sensitive ECNI-MS has enabled selected Parlar congeners to be detected and quantitated down to low parts-per-trillion (ppt) concentration levels in fish tissue extracts. The high sensitivity that organochlorine compounds exhibit under dissociative electron capture conditions is well documented (9,10).

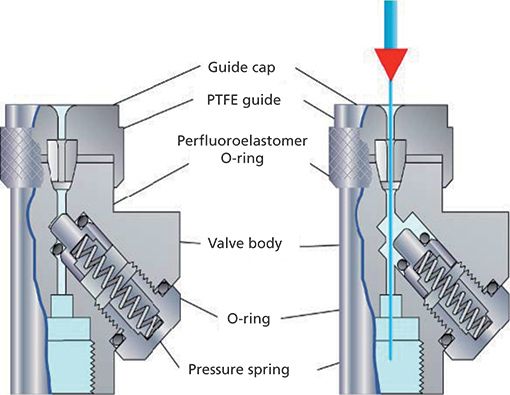
Figure 2: Schematic diagrams of the inner workings of the CIS-4 injector. (Courtesy of Gerstel Inc.)
A schematic of the injector is shown in Figure 1 and a detailed working illustration of the cooled septumless inlet is shown in Figure 2. Slowly injecting chemical analytes while the solvent is being vented serves to facilitate conditions for solvent removal. The application of SVSF injection enables an increase from the conventional 1-μL injection volume to a 10-μL volume to be made without chromatographic peak broadening. This volume reduction is accomplished by rapidly removing the solvent while depositing the analytes on a cooled inlet liner. Analytes are rapidly removed from the liner by ballistically heating the inlet under splitless injection conditions. A narrow band of analytes is swept onto the open tubular column. To optimize the LVI technique, if the extract volume is <10 μL, an unpacked baffle liner is recommended; a packed baffle liner using glass wool or beads is recommended if the extract volume is >10 μL. An unpacked baffle liner was used in this work. LVI does cause nonvolatiles to build up in the liner, and because of that potential problem the inlet liner needs to be checked frequently.

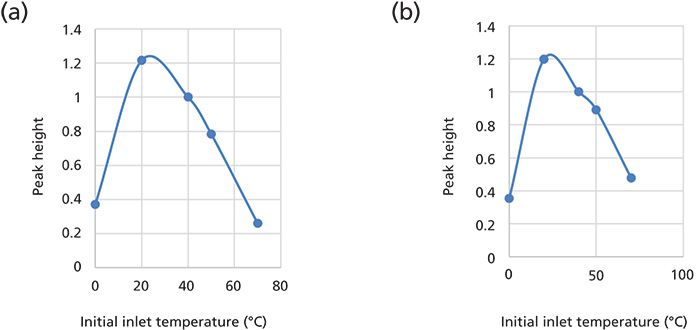
Figure 3: Experimental optimization of chromatographic instrument response versus injector initial inlet temperature for (a) P26 and (b) P50.
An important first consideration is where to set the initial inlet temperature. A rule of thumb suggests that the best results are obtained when the boiling point difference between the solvent and analyte is at least 150 °C (11). Experimentally, the initial inlet temperature should be set to 30 °C below the boiling point of the solvent. Fish tissue extracts injected into the LVI-SF inlet contain isooctane, whose boiling point is 99.3 °C (12). The effect of the initial inlet temperature on the chromatographic peak height (sensitivity) of selected Parlar congeners dissolved in isooctane was studied. Injecting only P26 while varying the injector temperature between 0 °C and 70 °C resulted in a maximum peak height as shown in Figure 3. This experiment was repeated using P50 and a similar maximum is also shown for it in Figure 3. Based on the inlet temperature that maximized the instrument response for P26 and for P50, a temperature of 40 °C was selected. This inlet temperature setting was used in all subsequent applications of SVSF. A screen capture for the SVSF parameters is shown in Figure 4. Analytical parameters for the autosampler are listed in Table II and the GC–ECNI-MS operating conditions were described in the text earlier.

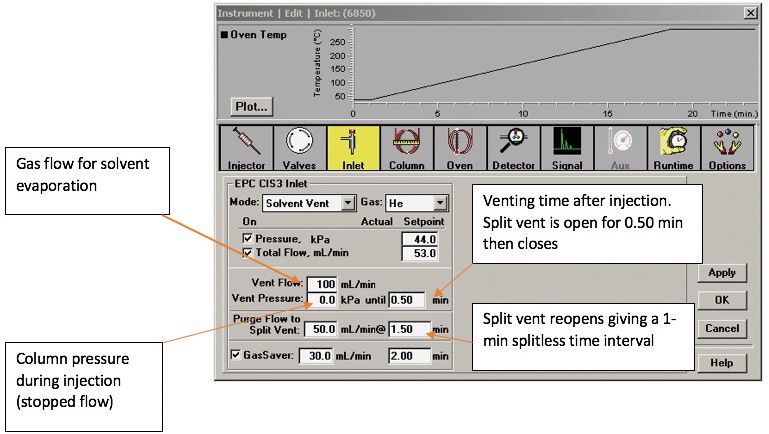
Figure 4: ChemStation/Maestro instrumental panel screen that shows how SFSV is implemented for the Gerstel CIS-4 injector. (Courtesy of Gerstel, Inc.)
The molecular structures of the Parlar congeners quantitated at ultratrace concentration levels (except for P44) are shown in Figure 5.

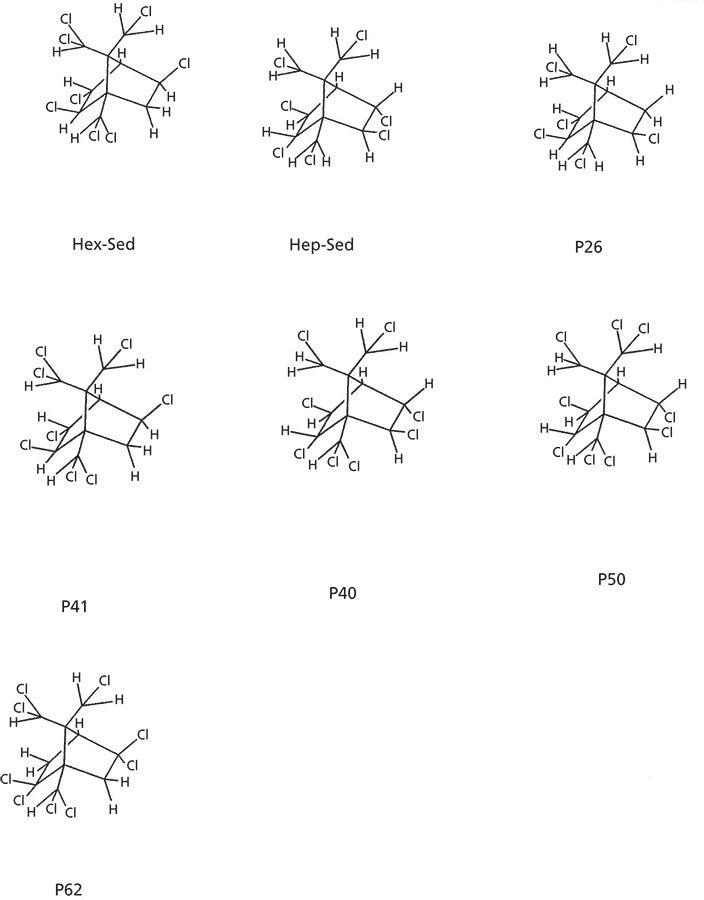
Figure 5: Molecular structures for the Parlar congeners of interest except for P44.
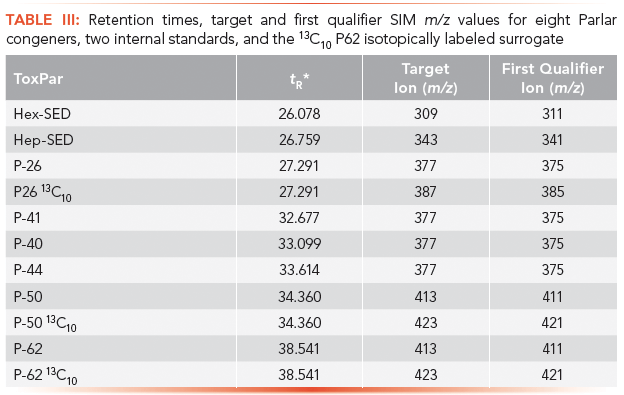
Table III lists the retention time, target and first qualifier selected ion monitoring (SIM) m/z ions for all Parlar congeners quantitated in fish tissue. The number of picograms of analyte injected for each P62 calibration standard is 2x the picograms injected for all other Parlar congeners quantitated. Hex-SED, Hep-SED, and P26 are quantitated against the 13C10 P26 internal standard while P41, P40, P44, P50, P62, and 13C10 P62 are quantitated against the 13C10 P50 internal standard. Retention times clearly show that isotopically labeled 13C compounds are coeluted with their unlabeled analogs.

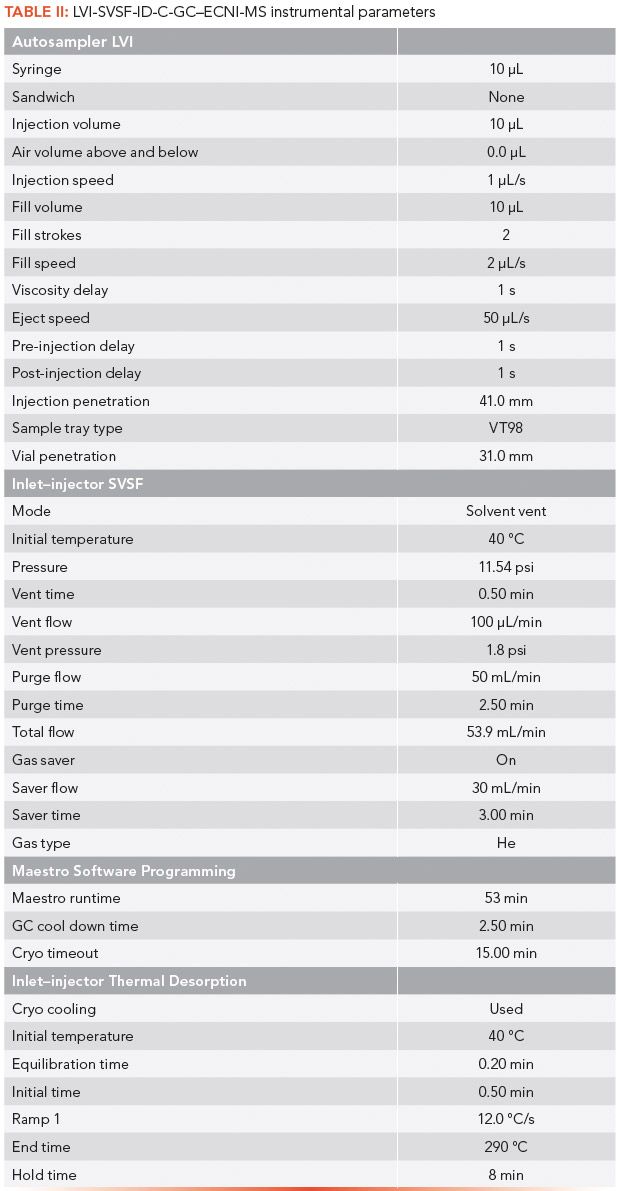
Figure 6 demonstrates that even a fast LVI-SVSF-ID-GC–ECNI-MS chromatogram can baseline separate the eight Parlar congeners in about 10 min. Refer to the text in the "Experimental" section for the temperature program used to quantitate the eight Parlar congeners whose results are shown in this article.

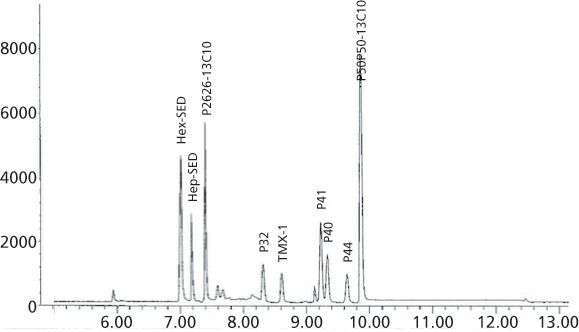
Figure 6: Typical LVI-SVSF-ID-GC-ECNI-MS chromatogram for the eight quantitated Parlar congeners.
Calibration and Establishing Reliable Instrument Detection Limits
The minimum criterion of r2 > 0.9900 was easily met for all Parlar congeners and for the surrogate 13C10 P62 using LVI-SF-GC–ECNI-MS. A new calibration for each Parlar congener and for 13C10 P62 was established for each new batch of sample extracts run. A comparison was made between calculating a limit of detection (LOD) based on a blank statistics model (3S0) and calculating an LOD based on a confidence band calibration statistics (CBCS) model. The results from this comparison are shown in Table IV. The method LOD, xMLOD, is calculated from the instrument LOD, xLOD, for each Parlar congener according to equation 1:


Equation 1 assumes an average lipid weight of 0.25 g and a final extract volume of 2.0 mL. A dilution factor of 2.5 is based on transfer of a 20-μL aliquot of fraction 2 extract and a 20-μL aliquot of fraction 3 extract to a vial that contains 10 μL of internal standard and yields a total extract volume of 50 μL.

For the 3S0 model, five replicate picograms-added values at each calibration level were found for each of the four lowest calibration levels that have detectable analytical peaks. The standard deviation for each of the four lowest calibration levels was plotted against the picograms injected. The y-intercept from a least squares fit to the data was obtained. The LOD was calculated from 3x the standard deviation in the y-intercept (3S0) and then interpolated to calculate the LOD.

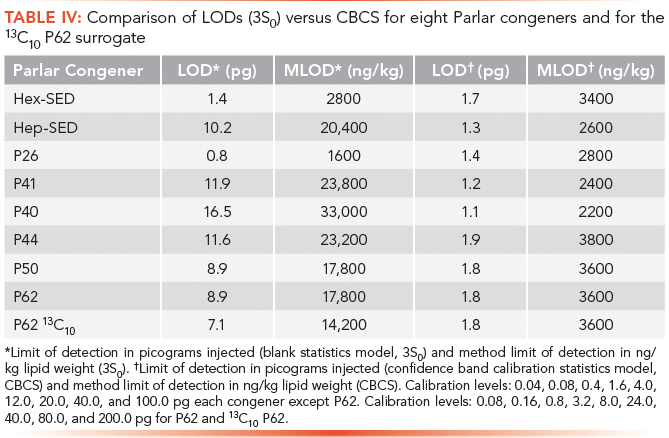
Table IV: Comparison of LODs (3S0) versus CBCS for eight Parlar congeners and for the 13C10 P62 surrogate
For the CBCS model, nine calibration points were used to calculate the weighted least squares regression line. Confidence intervals were also calculated. Where y critical is established on the y-axis (instrument response versus ratio of abundance of Parlar analyte to abundance of internal standard) at x = 0, serves to establish a critical response. From this critical response, the picograms injected (xcritical) and the number of picograms (xLOD) are found by interpolation (13,14).
It is evident from Table IV that applying the mathematics of CBCS resulted in lower and more consistent instrument detection limits across all the Parlar congeners considered.

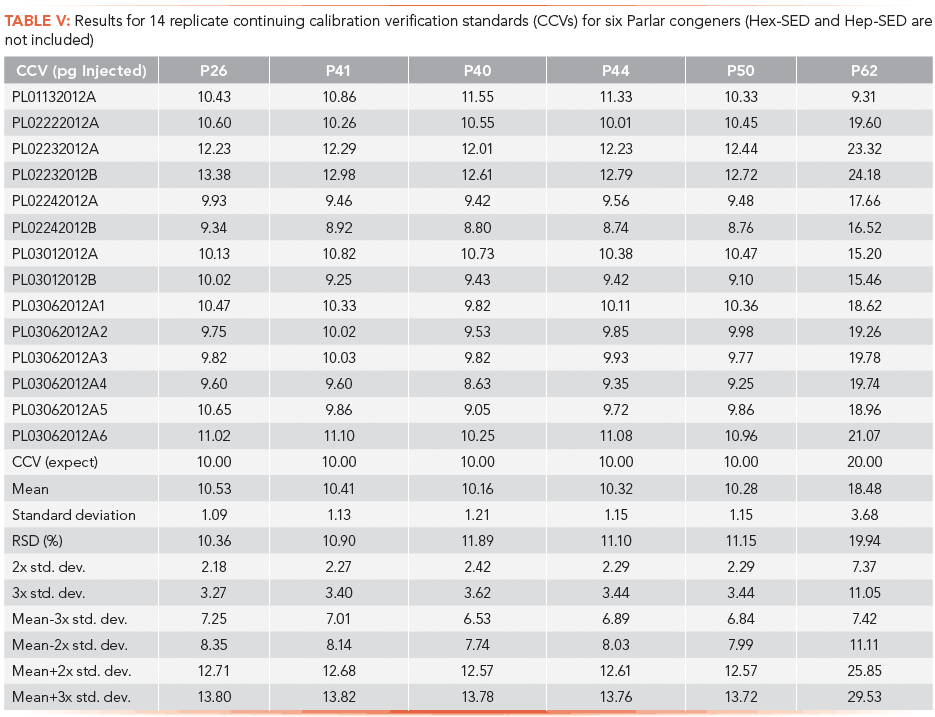
Quality Control
The method introduced here also met stringent quality control (QC) criteria. One continuing calibration verification (CCV) standard was included with each batch of samples run. An initial study investigated the precision over 14 replicate injections of the CCV standard. A mean standard deviation and relative standard deviation (RSD, expressed as a percent) were calculated as shown in Table V. The RSD was consistent across the first five Parlar congeners. Note that for P62, whose concentration levels were 2x those of the rest, the RSD reflected this difference. From the Table V statistical entries, QC charts were constructed. QC charts for P26 and P50 are presented in Figure 7 and show that instrument performance remained consistent.

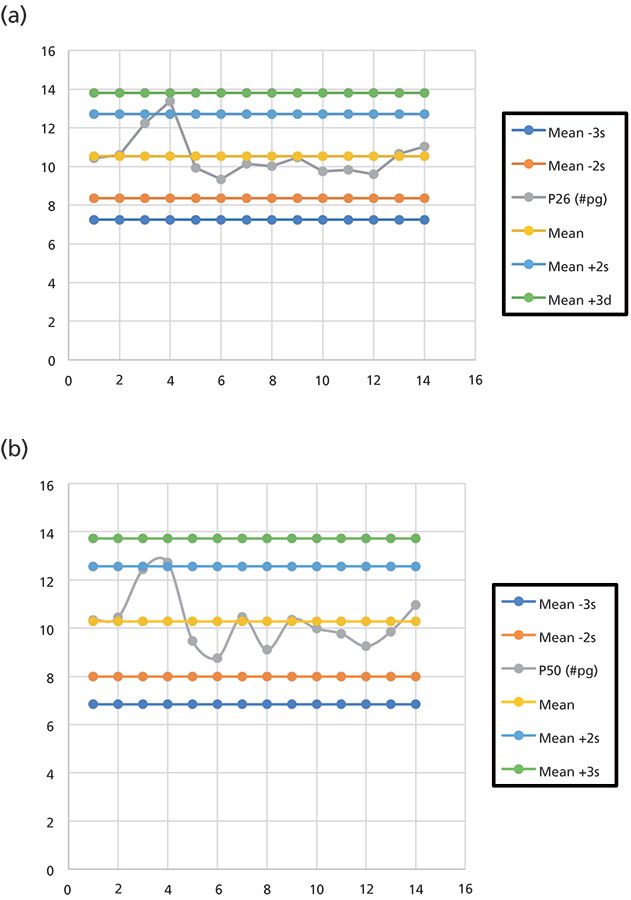
Figure 7: CCV QC charts for (a) P26 and (b) P50.
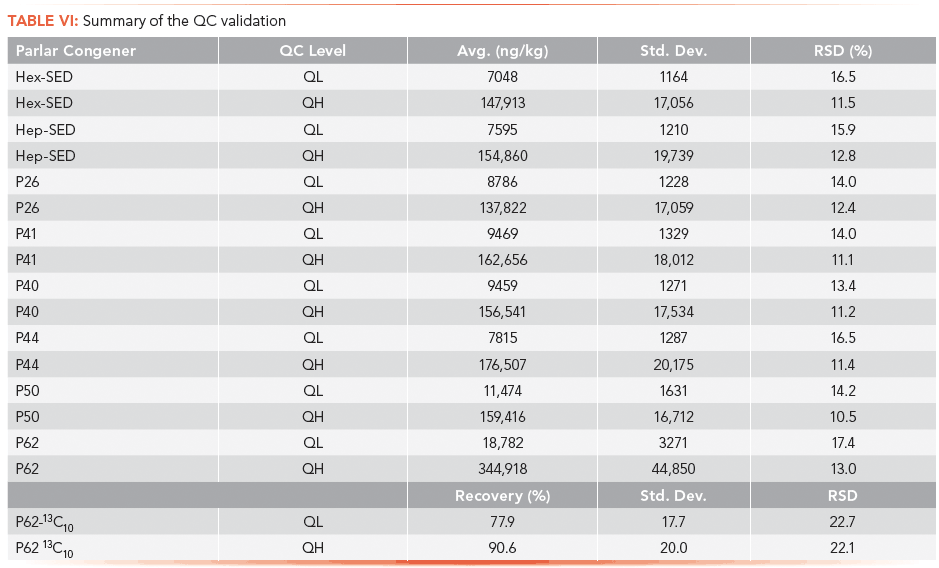
QC samples at a low concentration level (QL) and at a high concentration level (QH) were prepared by carefully spiking the eight Parlar congers and the 13C-labeled P62 surrogate into cod liver oil. Repeated injection of unspiked cod liver oil demonstrated its suitability as a blank substitute matrix for fish tissue. Results including a statistical evaluation of 23 replicate QC samples at QL and QH levels are summarized in Table VI. Equation 1 is used to report each QC result in nanograms per kilogram of fish lipid. RSD values over all Parlar congeners range from a low of 10.5% for P50 to a high of 17.4% for P62. A ~80% recovery for 13C P62 QL and a ~91% recovery for 13C P62 QH demonstrate good sample preparation efficiency.

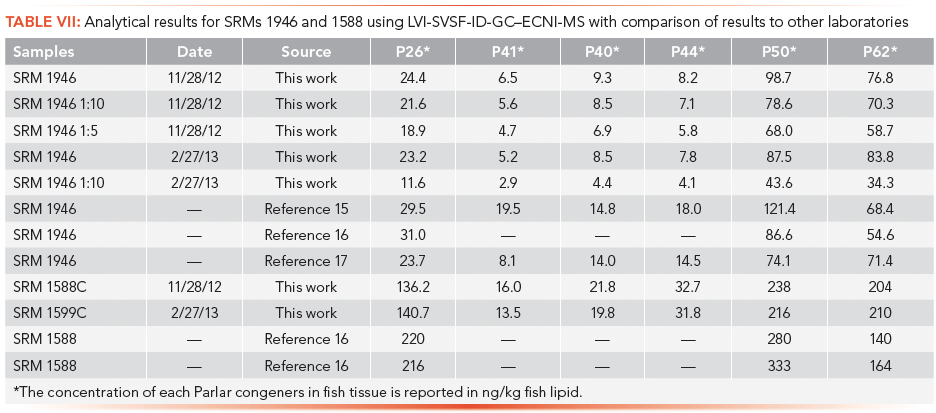
Our quantitative analysis of standard reference materials (SRMs) to determine the concentration of selected Parlar congeners in fish tissue and how these results compare to other laboratories are presented in Table VII. SRM 1946 and 1588 samples were analyzed and our results compared favorably with previously published results (15–17). Approximately 100 Great Lakes fish samples were analyzed using this modified EPA Method 8276 GC–MS approach.

Conclusion
This article has demonstrated that by combining LVI-SVSF-ID-GC–ECNI-MS with an efficient extraction, cleanup, and fractionation technique it is possible to extend instrument detection limits to the low parts-per-trillion concentration level for selected toxaphene Parlar congeners. A thorough QC study as applied to fish tissue has been presented.
Acknowledgments
David Elliott contributed significantly to sample prep while Michael O'Keefe meticulously prepared all the reference standards used. The efforts of the technical support teams from Gerstel and Agilent Technologies are greatly appreciated. This work originated from a request by the Michigan Department of Environmental Quality. Financial support was provided by: the Michigan Department of Community Health, Bureau of Laboratories, Lansing, Michigan, and the Department of Health and Human Services, Centers for Disease Control and Prevention, Public Health Emergency Preparedness.
References
(1) Method 8276: Toxaphene and Toxaphene Congeners by Gas Chromatography/Negative Ion Chemical Ionization Mass Spectrometry (U.S. Environmental Protection Agency, Washington, D.C., 2010, revisions in 2014, SW-846, EPA Office of Solid Waste).
(2) D. Hainzl, B. Burhenne, Jr., and H. Parlar, Anal. Bioanal Chem. 351, 271–285 (1995).
(3) S. Glassmeyer, K. Shanks, and R. Hites, Anal. Chem. 71, 1448–1453 (1999).
(4) D. Swackhamer, M. Charles, and R. Hites, Anal. Chem. 59, 913–917 (1987).
(5) P. Loconto, Trace Environmental Quantitative Analysis, 2nd edition (CRC Press, Taylor and Francis, Boca Raton, Florida, 2006), pp. 160–165.
(6) L. Needham, V..Burse, and H. Price, J. Assoc. Off. Anal. Chem. 64, 1131–1137 (1981).
(7) K. Smalling and K Maruya, J. Sep. Sci. 24, 104–108 (2001).
(8) K. Grob, Split and Splitless Injection in Capillary GC, 3rd Ed. (Hütig Buch Verlag, 1993), pp. 500–510.
(9) V. Ong and R. Hites, Mass Spectrom. Rev. 13, 259–283 (1994).
(10) J. Watson and O. Sparkman, Introduction to Mass Spectrometry, 4th Ed. (Wiley, Hoboken, New Jersey, 2007), pp. 464–469.
(11) "SVSF Using the CIS-4 inlet," product literature, Gerstel GmbH & Co. KG.
(12) The Merck Index, 13th Edition, M. O'Neil, Ed. (Merck & Co. Inc., 2001).
(13) P. Loconto, Am. Lab. 47(7), 34–39 (2015).
(14) J. Budge, D. MacTaggart, and S. Farwell, J. Chem.Edu. 76, 434–439 (1999).
(15) X. Xia, B. Crimmins, P. Hopke, J. Pagano, M. Milligan, and T. Holsen, Anal. Bioanal. Chem. 395, 457–463 (2009).
(16) J. Kuchklick and P. Helm, Anal. Bioanal. Chem. 385, 819–836 (2006).
(17) W. Lao, D. Tsukada, and K. Maruya, J.Chrom. A 1270, 262–268 (2012).
Paul R. Loconto, PhD, consults in analytical chemistry and in chemical education and is based in Okemos, Michigan. Direct correspondence to: locontop@att.net













Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.




This information is supplementary to the article “Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing”, which was published in the May 2025 issue of Current Trends in Mass Spectrometry.


