Quantifying Proteins by Mass Spectrometry
LCGC Europe
The quantification of proteins in a complex biological sample is an important and challenging task. Mass spectrometry (MS) is increasingly used for this purpose, not only to give a global survey of the components and their amounts, but also to precisely and accurately quantify specific target proteins.
The quantification of proteins in a complex biological sample is an important and challenging task. Mass spectrometry (MS) is increasingly used for this purpose, not only to give a global survey of the components and their amounts, but also to precisely and accurately quantify specific target proteins. Here, we review the essential elements of MS approaches to protein quantification and critically compare the available options.
Proteins are the most abundant macromolecules in biological systems. Together with their smaller relatives, peptides, they are polymers comprising amino-acid building blocks joined through amide bonds. In contrast to the repeating units of other biopolymers (for example, polysaccharides and polynucleotides), the constituent amino acids are diverse in their chemical and physical properties. Consequently, the polymers derived from them are also a complex, chemically and physically diverse ensemble.
This structural diversity lends itself to extensive functional diversity. Proteins serve as antibodies, enzymes, messengers, structural components, and transport or storage molecules. For that reason, the majority of drug targets are proteins. Significantly, the genetic machinery of the cell is tasked with synthesizing proteins. Accordingly, one might argue that much of each cell, and therefore any organism in toto — its structure, function, reproduction, repair, and regulation — relies on proteins. Understanding biology — function or dysfunction, health or disease — is therefore about defining and understanding proteins.
Protein identification and quantification are therefore the two central objectives of many biological and biomedical studies. Historically, these tasks were performed on purified proteins that were exhaustively sequenced (such as Edman) or quantified by the immuno-based western blot or enzyme-linked immunosorbent assays (ELISAs). Today, however, because of advances in mass spectrometry (MS) and the development of a set of global, protein-analysis tools that some call the "proteomics toolbox," improved analytical strategies have evolved, and the objectives of researchers have changed. Typically, investigators now aim to study biological entities at the "systems" level; that is, they seek to resolve and identify a multitude of proteins simultaneously in a single sample and to quantify each in relative or absolute terms (See definitions in Table 1).



Table 1: Some essential definitions.
Quantification is an important component of most studies. Defining differences or changes in protein abundances (or, more appropriately, the abundance of specific protein species including isoforms and post-translational variants) between two or more groups or states (such as control and test) is often at the heart of understanding function and regulation.
As the proteomics toolbox evolves, new approaches to protein quantification by MS are continually reported. These methods can be categorized into several major classes, all of which share features and performance characteristics. Furthermore, though some additional considerations are specific to protein quantification, it is also important to state that the process of quantification remains essentially the same, regardless of the nature of the analyte (that is, small molecules versus biopolymers). Consequently, the principles and practices that have guided the development and evaluation of quantitative methods (for example, replicate measurements to characterize the variance of a method) are no less applicable in this setting.
Non-MS Approaches to Protein Quantification
This column instalment focuses on MS methods for protein quantification because of their growing importance. Yet it is important to acknowledge the existence of other strategies and that, moreover, in certain settings those alternatives may be the methods of choice. For example, the mainstay for targeted protein quantification for almost 50 years has been the western blot immunoassay, in which antibodies are used to detect proteins transferred from polyacrylamide gels to nitrocellulose or polyvinylidene fluoride membranes. Refinements of the basic protocols yield detection limits in the attomolar range (1). Evolution of the principles underlying the western blot led to the development of the radioimmunoassay (RIA) and ELISA (2,3). In a clinical setting, specific proteins are commonly quantified by ELISA. Similarly, for several decades, quantification of multiple protein components in complex biological samples has relied on two-dimensional (2D) gel electrophoresis (discussed in some detail later). In addition, a growing array of multiplexed, selective capture methods, including aptamer and antibody arrays, are increasingly used for protein quantification. Numerous reviews discuss these and related strategies (4–9).
MS Approaches to Protein Quantification
An Overview of the Approaches: We categorize protein quantification by MS into several groups. These are outlined below, represented in Figure 1, and discussed in more detail in the sections that follow.

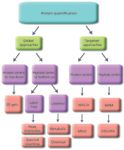
Figure 1: Diagrammatic representation of the common strategies for protein quantification and the relationships between them.
Global, System-Wide (Multicomponent) Strategies: Here, hundreds or thousands of proteins are compared in two or more samples and quantification is typically relative. Nevertheless, with modification to the basic method, absolute quantification is sometimes possible.
Protein-Centric or Top-Down Strategies: These approaches involve resolution of a complex mixture of proteins (for example, by 2D gel electrophoresis) and quantification in their intact form. The approach often involves differential radio- or chemical-labelling of proteins in distinct samples (such as, difference gel electrophoresis [DIGE]). After tagging, the samples are combined, the proteins are resolved in two dimensions, and the relative amounts of the tagged proteins are measured. Protein spots are excised from the gel, digested, and identified by MS by means of peptide mass maps (that is, peptide masses), peptide sequences (that is, tandem mass spectrometry [MS–MS]), or a combination of both techniques.
Peptide-Centric, Bottom-Up, or Shotgun Strategies: Here, a complex mixture of proteins, an extracted proteome, is digested to peptides. The peptides, which serve as surrogates of the original intact proteins, are then separated, quantified, and identified. These strategies are further subdivided into label-free strategies and labelled strategies, depending on whether a label is incorporated:
Label-Free Strategies:
- Isolate proteins in a sample → proteolysis → separate peptides → sequence peptides → identify protein → repeat procedure for additional samples. We then compare either the number of peptides recovered for each protein (that is, spectral counts) or relative abundances of specific peptide ions (that is, peptide peak intensities by liquid chromatography–mass spectrometry [LC–MS]) to quantify proteins.
- Labelled Strategies: This involves differential metabolic labelling with stable isotopes of proteins in two or more samples → combine samples → isolate total proteins → proteolysis → separate peptides → quantify differentially labelled peptides → sequence peptides and identify proteins (for example, stable isotope labelling by amino acids in cell culture [SILAC]).
- Differential chemical labelling with stable isotopes of proteins in two or more samples → combine samples → isolate total proteins → proteolysis → separate peptides → quantify differentially labelled peptides → sequence peptides and identify proteins (for example, isobaric tags for relative and absolute quantitation [iTRAC]). (Note the similarities of this approach to DIGE. The primary difference is that separation and quantification are performed on surrogate peptides, not intact proteins.)
Targeted (Single-Component or Several-Component) Quantification Strategies: In these approaches one or a few components are selectively isolated from a sample and quantified in relative or absolute terms. Approaches fall into two categories, top-down and bottom-up.
Top-Down Approach: Direct quantification by matrix-assisted laser desorption-ionization (MALDI) or protein isolation–concentration by an approach such as mass spectrometric immunoassay (MSIA, Thermo Fisher Scientific): Selective isolation of one or more proteins → determine protein abundances based on ion current. Protein identification or selectivity is derived from antibody and mass of target protein; amount is based on ratio of peak heights/areas for analyte and an internal standard. Absolute concentrations are determined referring to a calibration curve containing a fixed amount of internal standard (IS) and varying amounts of the intact target protein.
Bottom-Up Approach: Multiple-reaction monitoring (MRM) methods including stable isotope standard capture with anti-peptide antibodies (SISCAPA). Approaches selectively isolate target protein or proteins → digest proteins → quantify one or several peptides according to parent-ion (MS) or product-ion chromatograms (MS–MS).
General Considerations in Quantitative Proteomics by MS
An abundance of reviews discuss protein quantification by mass spectrometry, but most focus on instrumental considerations (10–13). Furthermore, most authors have almost exclusively focused on electrospray ionization (ESI)-based approaches and have neglected the findings of precise and sensitive intact-protein quantification by MALDI-based methods. Our focus is on the overarching steps in system-wide protein quantification.
Sources of Inaccuracy and Imprecision: Multiple sample-manipulation steps are common before instrumental analysis (for example, protein precipitation or isolation, fractionation, selective depletion and enrichment, proteolysis, and tagging and labelling reactions). Each step is a source of preanalytical sample variability that can compromise both precision and accuracy. For example, high-abundance proteins like albumin are sometimes removed from plasma samples by means of immunodepletion before analysis. However, that removal process introduces a risk of codepleting other components of interest because of nonspecific binding to both the antibodies used and to the albumin itself (by other sample components). Similarly, other steps such as protein precipitation and enzymatic digestion can introduce significant imprecision and inaccuracy because proteins are not recovered or digested quantitatively. Irreproducibility in other sample-handling steps, including chemical labelling, together with instrument perturbations (such as pressure and temperature fluctuations and tuning); the laboratory environment (for example, temperature and humidity); reagent variability; the presence of coeluted species or their levels; and analyte concentration can contribute to imprecision by altering the ionization process and thereby the measured signal intensity. Intensity comparisons are therefore compromised at a fundamental level. Studies that compare results across different analytical runs are most susceptible to these factors. Minimizing or carefully controlling key variables is critical. Yet even so, given the number of steps in the analysis and the number of species being measured, many potential sources of imprecision remain.
Validation of Proteomic Methods: It is important to remain mindful that the primary aim of any quantitative proteomics study is to provide timely, accurate, and reliable data that are fit for an intended purpose. Nevertheless, depending on the specific approach and the rigour with which the analysis is performed, the quality of the data will be variable and undefined. Quantifying thousands of components in a sample is a formidable challenge, to say the least. Defining specificity, linearity, accuracy, precision, range, detection limit, upper and lower limits of quantification, and robustness — all central considerations in the validation of a conventional quantitative analysis — is, given the scope of the task, empirically impossible for every protein species. Similarly, recovery and stability studies are not possible on each of the components. Consequently, in global proteomic studies, validation of the assay is typically perfunctory, and the resultant data are of uncertain and ill-defined reliability.
Although conventional validation is not practical, measures of precision and accuracy remain essential so that experimental findings can be put into context. Therefore, we must be confident that the measured differences are real and not merely an artifact of the method itself.
A method's assessed precision for a subset of analytes measured, at various concentrations, in one or more test samples and the derived data can be used to determine the method's suitability. These data can also help validate subsequent findings derived from the method. (14). Similarly, technical replicates (that is, repeat analyses of each of the samples in the study) provide additional support that a change is real, not an artifact of the analytical method itself. In the same vein, the issue of specificity must also be considered. Accurate quantification cannot be assumed on the basis of one — or even a few — peptides simply because a single peptide defines only a single segment of any protein, and modifications elsewhere in the molecule are missed (14,15). Quantification based on a peptide common to multiple, related forms will always lead to an overestimate of the amount of any single variant whereas quantification based on a unique peptide fails to "recognize" and quantify closely related variants of that protein, even if they are significantly more abundant. Precise and accurate quantification of a specific protein variant is therefore achievable only when the targeted peptide or peptides are derived from a single precursor protein or, in the case of protein-centric methods, in instances in which we can resolve and quantify the specific (intact) protein species without interferences.
The situation, however, is not as bad as it might seem at first. Because a common objective in proteomics is to compare groups — for example, disease versus control or control versus test — absolute levels are not (necessarily) important. Defining percent change (or difference) is the overarching objective. Therefore, it is possible to take advantage of differential (isotopic) tags and the exquisite selectivity of mass detection to compare two (or more) samples worked up and assessed in the same experimental run under identical conditions. For example, in a typical experiment all proteins in a sample (such as control) are labelled with a chemical tag; separately, all of the proteins in a second sample (such as disease) are labelled with an isotopic variant of the same tag. The samples are then mixed and treated as one. Thereafter, each tagged protein and its isotopic variant behave in an identical manner during sample handling. Yet because of their difference in mass, they can be specifically detected and quantified by MS. While this detection and quantification strategy markedly reduces variance in one sense, such an approach is limited to "A versus B" comparisons. Numerous modifications of the basic strategy have been developed and will be discussed in more detail later in this instalment.
Global or Proteome-Wide Protein-Centric Quantitative Tools
2D Gel-Based Methods: Using 2D gel-based methods is the most frequently adopted top-down strategy, and it is based on quantitative analyses of intact proteins resolved via 2D gel electrophoresis. Typically, the first step is protein separation by isoelectric focusing (IEF; first dimension [1D]) and then orthogonal separation of the proteins distributed on the 1D strip by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE; second dimension). Protein spots (that is, discrete species) are visualized by dyes, fluorophores, or radioactive labels tagged. (These can be visible or fluorescent post-electrophoretic dyes, or fluorophores or radioactive labels that are tagged to proteins before resolution. Examples include silver stain, Pro-Q Diamond, 2,4-dinitrophenylhydrazine, or Coommassie blue.) The quantity of each protein is assessed by measuring the spot's density using a customized software package. Two-dimensional gels provide excellent sensitivity, precision, and linearity over a wide dynamic range. Nevertheless, subsequent identification of proteins requires resecting, by hand or robot, the individual spots from the gel followed by MS (that is, a peptide mass map with or without additional MS–MS sequence information).
To minimize the influence of gel-to-gel variations, and to reduce the total number of gels required, variants of this general strategy involving differential labelling (or tagging) of the proteins in two or more samples have been developed (for example, with fluorophores or radioactivity labels). The objective of these approaches is to retain the physical properties of the proteins so that their mobility in each of the two dimensions of separation remains unchanged. Therefore, both can be run as a mixture on one gel, and each can be independently quantified, because of the tag.
The most common manifestation of this approach is DIGE (16). Here, three (or more) different protein-containing samples can be labelled with size-matched, charge-matched, spectrally resolvable fluorescent dyes (for example, Cy3, Cy5, and Cy2) before 2D gel electrophoresis. In this way, the number of gels to be resolved is reduced, precision is increased, and the time and cost of the comparison is also reduced. For example, running two samples on a single gel significantly increases reproducibility. Nevertheless, because three distinct "tags" are available, comparisons can also be made between multiple samples run across multiple gels by using a pooled internal standard (17). Composed of a mixture of all of the samples constituting the study, this standard is tagged and run on each gel. The pooled sample acts as an internal standard for every protein spot on each of the gels. As such, it is used to normalize all spots and to optimize inter-gel precision. Typically, reciprocal labelling (a dye-swap experiment) is performed to ensure the observed changes are not associated with dye-dependent interactions. Commercial software is used to detect differences and assign statistical confidence to them.
Critical Evaluation: With high precision, 2D gel electrophoresis and its variant, DIGE, allow relative abundance comparisons that detect modest changes of one to several thousand proteins in multiple samples. Provided they can be resolved, variant forms of the same protein (that is, protein variants or proteoforms) can also be independently quantified. Because each separation is visually represented, only proteins that differ in abundance need be resected from the gel and identified. Two-dimensional gel electrophoresis and DIGE have been successfully adopted by many investigators. Both methods have been thoroughly reviewed and have stood the test of time (18,19). However, the approach is cumbersome, labour-intensive, and difficult to fully automate. These limitations, especially the last, have dampened the enthusiasm of many to adopt 2D gel electrophoresis (and DIGE). While it is frequently suggested that the resolution of a 2D gel is limited, it is important to stress that no other approach rivals 2D gel electrophoresis for practical, intact protein separation. Comigration of multiple proteins to the same location on the gel is also often cited as a problem, but it is rarely an issue (20). Similarly, although large (>150 kDa) or small (<5 kDa) proteins are difficult to separate on gels, this difficulty is not a significant limitation because work-arounds are available, at least for peptides. DIGE is subject to variations in the extent of labels incorporated into the proteins, a potential source of variance in the measurements. Identifying proteins is sometimes problematic, especially with DIGE. Alignment problems can occur because identification requires running a separate (preparative) gel and then matching it to images for the set of analytical gels.
Other Top-Down Strategies: In this review, we forego discussion of instrument-intensive, top-down approaches simply because they are not yet practical for routine use. As alternatives to 2D gel, top-down proteomic approaches are, however, being developed by several groups. These alternatives involve introducing intact proteins into a mass spectrometer and fragmenting them directly. By working with the intact protein, complete sequence coverage is possible, and post-translational modifications (PTMs) are preserved. Until recently, however, top-down proteomic strategies were restricted to the analysis of purified proteins or simple mixtures. Over the last few years the approach has been extended to complex mixtures of proteins (21), but quantification remains a challenge nonetheless.
Global or Proteome-Wide Peptide-Centric Approaches
General Comments: Peptide-centric (bottom-up or shotgun) quantitative strategies are dominant because of their purported ability to quantify multiple components simultaneously in an automated or semi-automated manner. These strategies involve a common step: that is, site-specific cleavage of a mixture of isolated proteins (a proteome) to generate a substantially more complex mixture of peptides. Typically, peptide-centric applications utilize trypsin and quantify based on tryptic peptides. The selective cleavage of proteins to peptides is undertaken for these reasons: Peptides can be separated by LC better than proteins; most proteins generate one or more soluble peptides even if the antecedent protein is poorly soluble; peptides fragment better in a tandem mass spectrometer, giving spectra that can be sequenced; and peptides can be detected at much lower levels than their protein precursors. The peptides are then fractionated by LC and analyzed by MS–MS (14).
It is important to acknowledge, however, that all peptide-centric approaches to quantification are based on the assumption that when a protein is cleaved by a specific reagent, the reaction will go to completion, or at the very least, that the cleavage will be reproducible and predictable. Further, it is assumed that the target peptide or peptides are sufficient to define and selectively quantify the antecedent protein (that is, the target peptide is solely derived from a single antecedent protein). In practice, however, a target peptide or peptides may be degenerate and shared by multiple proteins. Through digestion, connectivity between the peptides and their antecedent protein is lost, a phenomenon referred to as the protein inference problem (22). In fact, multiple variants of a protein (variant protein species or proteoforms) are common, and unless peptides incorporating the specific, modified residue or residues (for example, the oxidized, reduced, nitrated, phosphorylated, glycosylated, or differentially "altered" amino acid) are targeted, quantification will be inaccurate. As a specific example, a single-point amino acid mutation may exist in a target protein, but if quantification is based on any tryptic peptide other than the one incorporating the modification, the variant will not be detected. Similarly, other variants of the precursor protein including truncated or alternatively spliced forms are often misidentified (15). On the other hand, if the focus is on identifying specific modifications, and the correct peptide is targeted, the peptide-centric approach offers advantages. The influence of a modification on mass is more evident at the level of the peptides than it is at the protein level because the percent change in mass is greater.
Relative quantification by peptide-centric methods can involve the separate analysis of multiple samples by MS and their subsequent comparison (for example, label-free methods). Alternatively, tags (such as isotopic tags or stable isotopes) can be incorporated into proteins or their proteolytic peptides. These cause a shift in mass of the labelled protein or peptides in the mass spectrum. Differentially labelled samples can be combined and analyzed together, determining differences in the peak intensities of the isotope pairs. These intensity differences correlate with differences in the abundance of their antecedent proteins.
With these over-arching issues in mind, the approach can be implemented in many, disparate ways. This review does not aim to be an encyclopedia of all available methods. Instead, it is a description of the basic divergent strategies and their strengths and weaknesses.
Label-Free Approaches: Label-free quantification is fast, cost-effective, and easy to implement. It is frequently used when stable isotope incorporation is impractical or cost-prohibitive. In these approaches, samples are analyzed separately and results from multiple runs are compared. The two main, label-free approaches each rely on proteolytic digestion of a sample followed by analysis by LC–MS or LC–MS–MS. Both strategies are used to make comparisons between two or more samples and to determine relative change in protein abundance (with the caveats noted above).
Spectral Counting: This is a practical, semi-quantitative measure of protein abundance in proteomic studies. Relative quantification by spectral counting compares the number of identified spectra associated with the same protein between different samples — that is, the total number of tandem mass spectra that match peptides to a particular protein as a measure of protein abundance within a complex mixture. The approach is based on the finding that increasing protein abundance results in an increase in protein-sequence coverage. Therefore, abundant proteins produce more MS–MS spectra than less-abundant proteins, and their antecedent peptides are sampled more often in fragment-ion scans than those derived from low-abundance proteins. However, important caveats are associated with this approach. Low-mass proteins (that is, those generating fewer fragments on proteolysis) are problematic; the dynamic range of the approach is limited; precision is poor and, consequently, small changes in protein abundances are difficult to determine. Several modifications of spectral counting, including the normalized spectral abundance factor (NSAF) approach (23), have also been reported. NSAF corrects for the fact that larger proteins yield more peptides on digestion than shorter proteins and also accounts for sample-to-sample variations associated with replicate analysis. A modified spectral counting strategy, absolute protein expression (APEX) profiling, has been used to measure the absolute protein concentration per cell after the application of several correction factors (24). Further refinements have also been made and the approach was recently reviewed (25).
Quantification Based on Peptide Peak Intensities as Determined by LC–MS: This approach to quantification is based on the observation that for a specific peptide separated and detected by LC–MS, the measured ion current increases with increasing concentration. Typically, ion chromatograms for each peptide of interest are extracted from an LC–MS run, and their peak areas are integrated over time. Peak areas for the same ion are then compared between different samples to give relative quantification; absolute amounts can also be calculated by reference to a calibration curve. Most often, ion currents derived from the intact, protonated, peptide ions are monitored, but product ions generated by MS–MS can also be used for quantification. (Product-ion detection increases selectivity, but at the expense of sensitivity.) The approach and computational strategies to manage the data have been reviewed (26).
While the relationship between the actual amount of protein and generated ion current holds true for standard samples of limited complexity, in practice, the analysis of digests of complex biological samples is far more problematic. For example, variations in temperature, pressure, sample preparation, injection volume, retention time, and the presence of coeluted species can significantly compromise precision. (Studies often extend over weeks — or even months — and changes in column, mobile phase, instrument condition, and calibration begin to manifest themselves.)
Critical Evaluation of Label-Free Approaches: Label-free approaches are inexpensive and simple to implement, but the old aphorism, "You get what you pay for" may apply. They allow "semi quantitative" comparisons between samples, but precision and reliability is low, in large part because without an internal standard the measured ion current is susceptible to many factors when it is measured in many separate runs. In a recent study by The Association of Biomolecular Resource Facilities (ABRF), data generated from digests of parallel lanes of gel-separated proteins were supplied to several groups. The task was to "identify" the proteins in the sample and determine which were elevated or reduced in intensity relative to the adjacent lane. Not surprisingly, participants failed to agree, and there was no evidence that either approach — spectral counting or intensity-based quantification — could reliably address this question (27).
Labelled Approaches to Global Protein Quantification
General Comments: These approaches uniquely tag the proteins in two or more samples with a stable-isotope tag. The tagging can be done metabolically (that is, by adding enriched amino acids into cell culture medium) or chemically (that is, by covalently binding a labelled moiety to the proteins). The samples are then combined and analyzed in a single run. Precision is markedly improved because two or more samples are compared within one run, but at the expense of the time, cost, and complexity of the overall analysis. The earlier in the analytical process the label is incorporated into the proteins, the better, but its (global) incorporation is far from straightforward.
Metabolic Labelling: In this approach to relative quantification, the proteins in two or more samples are labelled with isotopically distinct forms of amino acids by growing cells in enriched culture medium (for example, SILAC). The first report of this approach was by Ong and colleagues in 2002 (28). Typically, two populations of cells are grown in separate cultures, one in standard medium and the other in medium containing stable-isotope-labelled amino acids. After the samples are combined, the mass difference between proteins and their proteolytic peptides in the two populations can be detected by MS. The ratio of peak intensities in the mass spectrum for the labelled versus unlabelled forms reflect the relative protein abundances in the two samples. This approach delivers the highest precision because the label is incorporated before any analytical steps are undertaken, and it therefore accounts for sample handling biases through the whole analytical process. These advantages are in part offset by the cost of the strategy and the fact that the metabolic labelling approach is far from widely applicable. For example, it cannot be applied to the assessment of protein differences in biological fluids collected from human subjects.
Chemical Labelling: Because metabolic labelling is often not feasible, if a stable-isotope label is to be used, it must be introduced later in the workflow by chemically tagging peptides or proteins. Two basic strategies are commonly adopted, as discussed below.
Isotopic Labelling: The many variants of this general strategy all aim to add isotopic atoms or isotope-coded tags to peptides or proteins. Some are simple in concept whereas others combine multiple elements to react with differentially tagged and selectively recovered peptides. Once again, two separate samples are differentially tagged with isotopic labels, mixed, and analyzed. Labelling strategies include enzymatic labelling with 18O at the C-terminus of proteolytic peptides (29); global internal standard technology (GIST), in which deuterated acylating agents (for example, N-acetoxysuccinimide [NAS]) are used to label primary amino groups on digested peptides (30); and chemical labelling with formaldehyde in deuterated water, to label primary amines with deuterated methyl groups (31).
Commercial isotopic labelling reagents are also available. The best known commercial option is, perhaps, the isotope-coded affinity tag (ICAT) method (32). Several iterations of ICAT tags have emerged. The first generation of the reagent comprises three separate parts: a sulphydryl-reactive chemical crosslinking group, a linker, and a biotin entity. The reagent's two versions are an unlabelled form and a heavy form incorporating eight deuterium atoms. The sulphydryl-reactive group reacts with free thiols (that is, on cysteine residues); the biotin tag is used to selectively recover the tagged peptides (that is, through binding with avidin); and the linker provides the opportunity to differentially (mass) label two samples. Since not all proteins contain a cysteine residue, this approach is limited in that about 20% of the proteome may be missed. Furthermore, the incorporation of deuterium as the label is suboptimal because of a discernible isotope effect, which manifests itself as differences in retention time. A variant ICAT reagent incorporating 13C was reported several years later (33). A further refinement of the same basic strategy, isotope-coded protein labelling (ICPL), which tags lysine residues and the N-terminus on the intact proteins has also been reported (34). Importantly, ICPL allows the simultaneous comparison of three groups in a single experiment (that is, [2H7], [2H3], and [2H0] forms).
Isobaric Labelling: These are the most commonly used isotope tags. Isobaric labels are a set of matched reagents designed to react with peptides to give products of identical masses and chemical properties. Significantly, these products can incorporate carefully selected combinations of heavy and light isotopes. Although many different manifestations of isobaric labels exist, they all comprise the same basic components. Those components are a reactive moiety that functionalizes groups such as primary amines or cysteines, a mass reporter with a unique number of isotopic substitutions, and a mass normalizer with a unique mass that balances or equalizes the mass of the tag. Each different tag is designed to be of equal mass when bound to a peptide, but to cleave on collision-induced dissociation (CID) at a specific linker location, thereby delivering different-sized tags (reporters) that can be quantified independently. In a typical workflow, the proteins in various samples are isolated, enzymatically digested to peptides, and labelled with different isobaric tags. The separately labelled samples are then mixed and analyzed as one. On LC–MS analysis, the peptides are separated, fragmented to produce sequence-specific product ions, to determine sequence, and the abundances of the reporter tags are used to determine the relative amounts of the peptides in the original samples. Commercially available isobaric mass tags (for example, TMT and iTRAQ) allow the simultaneous analysis of multiple samples in one run (such as 4, 6, or 8 mass-unit differences).
Critical Evaluation: Labelled approaches to global protein quantification offer relatively high precision and multiplexing capability, and they suit many sample types. Nevertheless, they are based on the assumption that analytes will be quantitatively — or at least uniformly — labelled in all samples. Because these strategies are based on measuring proteolytic peptides as surrogates of proteins, the general considerations raised previously (that is, the assumption of complete digestion and selection of diagnostic peptides) apply to all of these methods.
Targeted Protein Quantification
Approaches to targeted protein quantification similarly can be divided into two distinct groups: those that detect and quantify intact proteins (typically by MALDI) and those that quantify one or more surrogate peptides derived from each protein (typically by LC–MS–MS).
Intact Protein Quantification: Although numerous investigators have demonstrated the ability of MALDI as a precise and accurate approach to protein quantification (35–37), the most powerful and widely adopted manifestation of MALDI protein quantification is the mass spectrometric immunoassay. Developed by Nelson and colleagues (38), this assay combines immunoaffinity column capture with MALDI detection and quantification to reduce the number of components in the sample. In contrast to a conventional ELISA, selectivity is achieved through both the antibody and mass-specific detection. In practice, the sample is passed through an immunoaffinity column; the column is washed, to remove other components; and the bound antigen is eluted directly onto a MALDI target, ready for MS.
For quantification by mass spectrometric immunoassay, fixed amounts of a modified form of the antigen, or a similar protein, are typically added to the sample early in the process, as an internal standard. Absolute quantification is possible by reference to a calibration curve prepared and run in concert with the samples. Mass spectrometric immunoassay offers high-throughput protein quantification. It is important to note that it can also provide details about PTMs and genetic variants. In fact, not only is it possible to identify protein heterogeneity, but the variant forms of the same protein can also be independently quantified. Several different antibodies can be combined in a single column, to allow multiplexed antigen quantification. Although mass spectrometric immunoassay is most commonly combined with MALDI, ESI-based methods have also been developed (39).
Multiple Reaction Monitoring Approaches: Targeted quantification of proteins following their proteolysis to constituent peptides has increasingly become a routine task (40). With a few significant modifications, the process essentially follows the same strategy as that described earlier for "label-free methods." First, the target peptides are monitored in MRM mode. Then stable, isotope-labelled versions of the target proteolytic peptides are typically added as an internal standard (though the approach has also been used without incorporating an isotopic internal standard) (41). Monitoring more than one MS–MS transition for each target species provides a powerful approach to quantify a predetermined set of proteins for multiple samples, and it can potentially offer precise and accurate absolute quantification. Each target protein is cleaved to yield peptides, many of which have a unique sequence (that is, signature, or "proteotypic" peptides). A stable, isotope-labelled version of each signature peptide, designed to be identical to the tryptic peptides generated during digestion, is added at a fixed concentration to each sample, to serve as an internal standard. Because the labelled peptides are coeluted with the target peptide, the internal standards enter the mass spectrometer at the same time as the sample-derived peptides, and therefore they can be concomitantly analyzed by MS–MS. Typically, this approach is performed on a triple-quadrupole mass spectrometer or a hybrid (for example, a quadrupole combined with time-of-flight [TOF] or orbital ion trap analyzer). The target peptide concentration is determined by measuring its observed signal response relative to that of the stable-isotope internal standard. Absolute concentrations can be calculated referring to a calibration curve prepared at the same time. (Calibration curves must be generated for each target peptide in the sample.) With thoughtful selection of the target peptides, it is possible to quantify a specific protein or even a modified form of that protein. No antibody is required, and the process can be performed simultaneously on multiple — even hundreds of — peptides. Therefore, multiple proteins can be quantified in a single LC–MS–MS run. A variant of this process, known as parallel reaction monitoring (PRM), allows simultaneous monitoring of all product ions of a target peptide, rather than only a few predetermined transitions (42).
Critical Considerations: Selection of the specific peptides is a central issue because they should be diagnostic of the full target protein; use of just one or two peptides can lead to overestimations of proteins (15). (As discussed previously, target peptides could be common to known variants of the same protein.) Other important considerations address the possibility of incomplete digestion and the fact that sensitivity can be limited in the case of low-abundance proteins without an isolation or enrichment step. A major benefit of this strategy is that cost-effective, precise, and accurate analysis is possible without access to immunoreagents. Yet the approach can prove costly because of the requirement for multiple, stable, isotope-labelled peptides for each target protein. It can also prove time-consuming, because of the need to analyze the potentially complex MRM data.
Stable-Isotope Standard Capture with Antipeptide Antibodies
Stable-isotope standard capture with antipeptide antibodies (SISCAPA) is essentially the same procedure as that described above, except that it incorporates a specific, antipeptide antibody capture step for the signature peptide and its companion internal standard (43). The additional step enriches the sample for the target peptide and stable isotope standard. At the same time, it provides an opportunity to deplete the sample of interferents, including other peptides generated during the digestion. Importantly, because the internal standard is a perfect mimic of the target peptide, the peptide-to-internal standard ratio is preserved throughout the workup process. Extensive washing can be undertaken, to remove other peptides and clean the sample, without introducing additional variability in the results. The sample is then resolved by a short reversed-phase LC separation and analyzed by LC–MS–MS. Ions characteristic of the target peptide and its corresponding internal standard are monitored in MRM mode. From the signature peptide-to-internal standard ratio, the concentration of the peptide can be calculated by reference to a calibration curve.
Critical Evaluation: Antibody quality is important. So, too, is the selection of the specific peptides. The use of a single peptide is fraught with the problems discussed above. The limit of detection is improved because of the opportunity to trap and enrich the target peptides, but the cost and complexity of the approach is increased by the inclusion of this step.
Conclusions
MS quantification is not a trivial undertaking, even for small molecules. When the task at hand is the quantification of hundreds to thousands of proteins in a single sample, the complexity of the analysis is even greater. Shortcuts compromise the process and lead to unsatisfactory and irreproducible results. Nevertheless, with proper care, reflection upon sources of variance, and attention to generating reproducible results, multicomponent protein quantification fit for specific purposes are possible.
Acknowledgement
We thank Professor Jens R. Coorssen of the School of Medicine at the University of Western Sydney, NSW, Australia, for helpful comments.
Mark W. Duncan, PhD, is active in the application of mass spectrometry to the development of biomarkers for disease diagnosis and patient management. He currently works both as an academic in the School of Medicine at the University of Colorado, USA, and as a research scientist at Biodesix Inc., in Boulder, Colorado. He also holds a visiting appointment at King Saud University in Riyadh, Saudi Arabia.
Alfred L. Yergey, PhD, obtained a B.S in chemistry from Muhlenberg College and his PhD in chemistry from The Pennsylvania State University. He completed a postdoctoral fellowship in the Department of Chemistry at Rice University. He joined the staff of the National Institute of Child Health and Human Development (NICHD), NIH in 1977 from which he retired in 2012 as Head of the Section of Metabolic Analysis and Mass Spectrometry and Director of the Institute's Mass Spectrometry Facility. Yergey was appointed as an NIH Scientist Emeritus upon retirement and maintains an active research program within the NICHD Mass Spectrometry Facility.
P. Jane Gale, PhD, is currently Director of Educational Services at Waters Corporation. She has spent her career working in the field of mass spectrometry, first at RCA Laboratories and later at Bristol-Myers Squibb, where she was responsible for overseeing the development of quantitative bioanalytical assays to support clinical trials. She is a long-time member of the American Society for Mass Spectrometry (ASMS) and, together with Drs. Duncan and Yergey, created and taught the course in Quantitative Analysis given by the society at its annual conference for nearly a decade.
"MS — The Practical Art" Editor Kate Yu joined Waters in Milford, Massachusetts, USA, in 1998. She has a wealth of experience in applying LC–MS technologies to various application fields such as metabolite identification, metabolomics, quantitative bioanalysis, natural products, and environmental applications. Direct correspondence about this column should be addressed to "MS – The Practical Art", LCGC Europe, Honeycomb West, Chester Business Park, Chester, CH4 9QH, UK, or e-mail the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com
References
(1) J.R. Coorssen et al., Anal. Biochem. 307(1), 54–62 (2002).
(2) E. Engvall, K. Jonsson, and P. Perlmann, Biochim. Biophys. Acta 251(3), 427–434 (1971).
(3) E. Engvall and P. Perlmann, Immunochemistry 8(9), 871–874 (1971).
(4) M. Cretich, F. Damin, and M. Chiari, Analyst 139(3), 528–542 (2014).
(5) D.A. Hall, J. Ptacek, and M. Snyder, Mech. Ageing Dev. 128(1), 161–167 (2007).
(6) T. Joos, Expert Rev. Proteomics 1(1), 1–3 (2004).
(7) D. Stoll et al., Front Biosci. 7, c13–32 (2002).
(8) S.C. Tao, C.S. Chen, and H. Zhu, Comb. Chem. High Throughput Screen 10(8), 706–718 (2007).
(9) M.F. Templin et al., Trends Biotechnol. 20(4), 160–166 (2002).
(10) M. Nikolov, C. Schmidt, and H. Urlaub, Methods Mol. Biol. 893, 85–100 (2012).
(11) M. Bantscheff et al., Anal. Bioanal. Chem. 404(4), 939–965 (2012).
(12) K.M. Coombs, Expert Rev. Proteomics 8(5), 659–677 (2011).
(13) W. Zhu, J.W. Smith, and C.M. Huang, J. Biomed. Biotechnol. 2010, 840518 (2010).
(14) M.W. Duncan, R. Aebersold, and R.M. Caprioli, Nat. Biotechnol. 28(7), 659–664 (2010).
(15) M.W. Duncan, A.L. Yergey, and S.D. Patterson, Proteomics 9(5), 1124–1127 (2009).
(16) M. Unlu, M.E. Morgan, and J.S. Minden, Electrophoresis 18(11), 2071–2077 (1997).
(17) A. Alban et al., Proteomics 3(1), 36–44 (2003).
(18) M.W. Duncan and S.W. Hunsucker, Exp. Biol. Med. (Maywood) 230(11), 808–817 (2005).
(19) J.F. Timms and R. Cramer, Proteomics 8(23–24), 4886–4897 (2008).
(20) S.W. Hunsucker and M.W. Duncan, Proteomics 6(5), 1374–1375 (2006).
(21) J.C. Tran et al., Nature 480(7376), 254–258 (2011).
(22) A.I. Nesvizhskii and R. Aebersold, Mol. Cell. Proteomics 4(10), 1419–1440 (2005).
(23) B. Zybailov et al., J. Proteome Res. 5(9), 2339–2347 (2006).
(24) P. Lu et al., Nat. Biotechnol. 25(1), 117–124 (2007).
(25) L. Arike and L. Peil, Methods Mol. Biol. 1156, 213–222 (2014).
(26) M.M. Matzke et al., Proteomics 13(3–4), 493–503 (2013).
(27) D.B. Friedman et al., Proteomics 11(8), 1371–1381 (2011).
(28) S.E. Ong et al., Mol. Cell Proteomics 1(5), 376–386 (2002).
(29) O.A. Mirgorodskaya et al., Rapid Commun. Mass Spectrom. 14(14), 1226–1232 (2000).
(30) A. Chakraborty and F.E. Regnier, J. Chromatogr. A 949(1–2), 173–184 (2002).
(31) J.L. Hsu et al., Anal. Chem. 75(24), 6843–6852 (2003).
(32) S.P. Gygi et al., Nat. Biotechnol. 17(10), 994–999 (1999).
(33) E.C. Yi et al., Proteomics 5(2), 380–387 (2005).
(34) A. Schmidt, J. Kellermann, and F. Lottspeich, Proteomics 5(1), 4–15 (2005).
(35) X. Tang et al., Anal. Chem. 68(21), 3740–3745 (1996).
(36) M. Bucknall, K.Y.C. Fung, and M.W. Duncan, J. Am. Soc. Mass Spectrom. 13(9), 1015–1027 (2002).
(37) A.S. Benk and C. Roesli, Anal. Bioanal. Chem. 404(4), 1039–1056 (2012).
(38) R.W. Nelson et al., Anal. Chem. 67(7), 1153–1158 (1995).
(39) S. Peterman et al., Proteomics 14(12), 1445–1456 (2014).
(40) A.J. Percy et al., Biochim. Biophys. Acta 1844(5), 917–926 (2014).
(41) W. Zhi, M. Wang, and J.X. She, Rapid Commun. Mass Spectrom. 25(11), 1583–1588 (2011).
(42) A.C. Peterson et al., Mol. Cell. Proteomics 11(11), 1475–1488 (2012).
(43) N.L. Andersonet al., J. Proteome Res. 3(2), 235–244 (2004).














Free Poster: NDSRI Risk Assessment and Trace-Level Analysis of N-Nitrosamines
April 25th 2025With increasing concern over genotoxic nitrosamine contaminants, regulatory bodies like the FDA and EMA have introduced strict guidelines following several high-profile drug recalls. This poster showcases a case study where LGC and Waters developed a UPLC/MS/MS method for quantifying trace levels of N-nitroso-sertraline in sertraline using Waters mass spectrometry and LGC reference standards.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.
Using the Carcinogenic Potency Categorisation Approach (CPCA) to Classify N-nitrosamine Impurities
April 25th 2025Learn how to manage nitrosamine impurities in pharmaceuticals with our free infographic. Discover how the CPCA approach establishes acceptable intake limits and guides the selection of NDSRI reference samples. Stay compliant and ensure safety with our ISO-accredited standards.

