Pressure-driven Chromatography in Perfectly Ordered Pillar Array Columns
LCGC Europe
his article reveals the first liquid chromatography (LC) separations performed on a microfabricated pillar array column under pressure-driven conditions. The pillars were non-porous and produced using a Bosch-type deep reactive ion etch (DRIE) to pattern the surface of a silicon wafer and had a diameter of approximately 5 μm. Two different packing densities were compared: one similar to the packing density of a packed bed (external porosity of approximately 49%) and one similar to the packing density of monolithic columns (external porosity of approximately 70%).
This article reveals the first liquid chromatography (LC) separations performed on a microfabricated pillar array column under pressure-driven conditions. The pillars were non-porous and produced using a Bosch-type deep reactive ion etch (DRIE) to pattern the surface of a silicon wafer and had a diameter of approximately 5 μm. Two different packing densities were compared: one similar to the packing density of a packed bed (external porosity of approximately 49%) and one similar to the packing density of monolithic columns (external porosity of approximately 70%).
The most densely packed pillar array yielded plate heights between 0.9–4.9 μm (depending on the degree of retention of the employed coumarin test compounds). This is considerably smaller than for a packed bed column filled with particles with the same diameter, demonstrating the large potential advantages of ordered pillar array columns compared with the much more random structure of packed beds. The main obstacle to developing this new type of column technology at the moment is the lack of reproducible coating procedures.
The quality of a chromatographic separation appears to be extremely sensitive to the fine structure of the device it is performed in. This sensitivity is orders of magnitude larger than other basic reaction and separation processes such as extraction, centrifugation and heterogeneously catalysed reactions. Even for columns packed with highly monodispersed particles, small differences in packing heterogeneity already lead to a measurable effect on band broadening. In their 1998 landmark paper, Bing He and Fred Regnier suggested the use of microfabrication techniques from the microelectronics industry to produce chromatographic beds that are perfectly homogeneous.1
This groundbreaking new approach to column manufacturing also has potential to optimize particle shape and inter-particle distance as well as radically solve the packing reproducibility problems of packed bed columns.2



The potential of a perfectly ordered chromatographic bed by is high. Computational fluid dynamics (CFD) simulations of the flow and diffusion of retained species in ordered porous media demonstrated that the best possible commercial high performance liquid chromatography (HPLC) columns currently available yield minimal plate heights that are more than a factor of two larger than necessary, even if filled with particle batches with a very narrow particle size distribution.3–5
This was in agreement with earlier calculations and predictions made by, amongst others, John Knox.6 If the particles in a column could be positioned on the connection points of some imaginary perfectly ordered grid, minimal plate heights smaller than one particle diameter would be possible.3–5 Traditional column packing technologies are known to be incapable of yielding plate heights that are smaller than twice the size of the particle diameter.7,8
This translates into a doubling of the plate number without having to change any other parameter of the column packing. Moreover, since the minimum time needed to achieve a given separation resolution is related to the square of the plate height,8,15 a perfectly ordered equivalent of a packed column would allow for a four-fold reduction of the analysis time (whilst keeping the same resolution and comparing particles with the same intraparticle porosity).
The gain could even be made larger if the distance between the pillars was optimized (read enlarged). In this instance, gains up to a factor of 10 in analysis time would be achievable, provided columns with an optimized length were used.3
However, Regnier's group mainly focused on electro-driven capillary electrochromatography (CEC) techniques when exploring the microfabricated column concept.9,10 This was partly because this was pretty much in vogue at that time, but also because the electro-driven flow is less sensitive to slight local differences in pillar size than pressure-driven flow. The microfabrication technology they had at their disposal at that time was not really perfect and yielded pillars that were not as uniformly positioned and sized as needed. They apparently also experienced problems with the sealing between the pillar array and the cover glass. In a pressure-driven mode, the existence of a gap between the pillars and the "roof" wall creates a preferential flow path, or at least an additional dead volume, which can lead to strongly increased band broadening. When a system of subsequently bifurcating distribution channels was used, slight differences in flow resistance were observed between the different channel branches (see Figure 6 of He et al.1). These small differences were inevitable with the etching technology that was available at that time.
With the advent of new, more accurate etching technologies, such as the Bosch process,11–14 it is now possible to etch micro-pillar arrays with a much higher spatial resolution. Another advantage of the Bosch process is that it produces pillars that are much higher than previously possible while retaining a uniform diameter over the entire height. This prevents the creation of a velocity gradient between the top and bottom wall of the pillar array that would otherwise be detrimental for the separation resolution.
In the present study, we attempt to bring Regnier's microfabricated column idea to work under pressure-driven flow conditions. This work occurs partly in collaboration with the group of Peter Schoenmakers and Wim Kok of the Vrije Universiteit Amsterdam. The first results of this work were recently published in reference 14. It was shown there that, under non-retained conditions, a minimal plate height (H) as small as 2 μm can be obtained on a bed of circular pillars with a particle diameter (dp) of 10 μm. In reduced plate height (h) terms (calculated from h = H/dp) this corresponds to a value of h = 0.2, (i.e., 10 times smaller than the value of h = 2), which is generally considered as the performance limit of a packed column.7,8,15
This article describes the first reported LC separations performed on a microfabricated pillar array column under pressure-driven conditions. The base material of the chips is silicon but the surface of the pillars can easily be converted into silicon dioxide (in fact this already occurs spontaneously if simply exposed to the atmosphere). Consequently, the same silanol-based derivatization protocols as those traditionally employed for silica-type materials can be used to apply a retentive monolayer of C8- or C18-molecules on the outer pillar surface.
It was, therefore, straightforward to test the microfabricated columns under reversed-phase LC conditions. Channels with two different packing densities were considered: one type had a packing density slightly larger than a packed bed (which have an external porosity ε of approximately 49%), while the other type had a packing density that is similar to that of monolithic columns (which have an external porosity ε of approximately 70 %).
Experimental
Channel fabrication: The pillar channels were defined in a silicon–glass sandwich. First, a 100 mm diameter (100) silicon wafer (p-type, 5–10 Ω cm resistivity) was thermally (dry) oxidized at 1100 °C until 200 nm silicon oxide was formed (Amtech tempress omega junior). Then, normal ultraviolet (UV) photolithography (photoresist: Olin 907–12) was used to define the pillar array. Subsequently, the exposed silicon oxide was dry etched (Adixen AMS100DE, France), after which the resist was removed by oxygen plasma and nitric acid. Another lithography step was then used to define the inlet and outlet channels.
The exposed silicon oxide was then etched and a Bosch-type deep reactive ion etch (Adixen AMS100SE) was used to make these channels 90 μm deep. After stripping the resist, another 10 μm was Bosch-etched into the exposed silicon, leaving pillars of 10 μm height and supply channels with a total depth of 100 μm. To remove the fluorocarbons, the wafers were set in an oxide furnace at 800 °C for 30 min, after which the wafers were cleaned in nitric acid and dipped in hydrogen fluoride. The top of the channels was formed by a 100 mm Pyrex wafer. On the silicon wafer, through-holes were first defined by photolithography on a dry resist foil (Ordyl BF410). The exposed glass was subsequently powder blasted using 30 μm alumina particles. After stripping the foil, the substrates were anodically bonded (voltage ramped to a maximum of 1000 V at 400 °C, on an EVG EV–501 wafer bonder (EVGroup Inc., Austria).
Each pillar was positioned on the grid points of a connected array of equilateral triangles. In a first channel, the pillars had a mean diameter of 4.45 μm, and the side of each triangle (determining the distance between the centre points of two adjacent cylinders) was 5.93 μm. With these data, it can be calculated that the external porosity, which is equal to the relative interstitial volume (ISV), of this channel is equal to an ε value of 49% (i.e., of the same order as in a packed bed column). In a second channel, the pillars had a mean diameter of 4.68 μm, while the distance between the centre points of two adjacent cylinders was equal to 8.22 μm. In this instance ε was 70%.
Chip coating procedure: To enable reversed phase LC separations, the mantle surface of the microfabricated pillars was covalenty coated with a monolayer of hydrophobic C18 chains. However, prior to this bonding reaction, the chip was first pretreated to increase the efficiency of the bonding. Both the pretreatment and the bonding step were performed using a commercial nano-flow HPLC system (Agilent Technologies 1100 series, Germany). The connection between the pump and the chip was established by means of a home-made set-up. In this set-up, the chip was sandwiched between two holders, of which the top one (made of Delrin) was perforated with drilled through-holes that were compatible with commercially available nanoport connectors (Achrom, Belgium). The tubing used to connect the pump and the chip consisted of fused silica capillaries (Achrom) with an i.d. of 150 μm.
The pretreatment procedure consisted of the following steps. The chip was first rinsed with HPLC-grade methanol (Fluka, Belgium) for 2 hours at a flow rate of 2 μL/s. Afterwards, polyetheretherketone (PEEK) tubing (i.d. = 1 mm) with a length of 1.5 m was filled with 0.1 M sodium hydroxide (Fluka) using a commercial syringe and subsequently inserted between the pump and chip by applying two capillary PEEK-tubing adaptors (Achrom) at both ends of the tubing. With the pump as the driving force, this solution was chased through the chip for 2 hours at a flow-rate of 2 μL/min. This step was applied to activate the internal surfaces of the chip to produce a high density of silanol groups, which is necessary for the bonding reaction. These activated silanol groups originate from the native oxide layer that is spontaneously formed on the silicon surface of the pillars. Afterwards, the chip was flushed with 0.1 M nitric acid (Fluka) in a similar way.
The same strategy was used for the introduction of the coating solution. A 20% (v/v) solution of octadecyldimethylchlorosilane (Sigma-Aldrich, Belgium) in HPLC-grade toluene (Sigma-Aldrich) was prepared and introduced into the PEEK-tubing. To increase the kinetics of the bonding reaction, a home-made heating block (Gefran, Belgium) was brought in physical contact with the chip through an aperture machined in the aluminium bottom holder of the set-up. While the chip was kept at a temperature of 50 °C, the coating solution was chased with toluene through the column at a flow-rate of 2 μL/min for 5 hours. After 5 hours the column was washed with toluene and methanol for 3 hours, respectively.
System hardware and separation procedures: The basic principle of the employed injection procedure (Figure 2) has been described previously by Chmela et al. and is based on the presence of an injection zone devoid of pillars (200 μm wide in the present situation).16 The sample injection consisted of two consecutive steps: the definition of a sample plug with a certain defined axial width [step 1, Figure 2(a) and (b)], followed by the transportation of this plug along the column by the mobile phase flow [step 2, Figure 2(c) and (d)]. During the first step of the injection, the outlet ports for channels 1 and 4 are closed so that the only convective flow that can be generated is from port 3 to port 2. As a result, a rectangular sample plug is defined at the injection zone. In the second injection step, ports 1 and 4 are opened while ports 2 and 3 are partially closed. An additional description of the injection procedure is given in the caption of Figure 2.
A home-made set-up with pressurized vessels, controlled by two pressure controllers, was used to provide the required pressure for the flow generation in the two steps of the injection process (Figure 3). The sample reservoir and the mobile-phase reservoir consisted of two different home-made stainless steel vessels. These vessels were independently connected to two different pressure controllers (Bronkhorst, The Netherlands), fed by a nitrogen gas bottle, that allowed the control of the pressure in the vessels with a precision of the magnitude of millibars. This approach allowed applying two different independent pressure settings, one for the sample injection and one for the mobile phase flow. Wide capillaries (i.d. = 150 μm, Achrom) were used for the connection between the pressurized vessels and the chip, so that the pressure readout of the pressure controllers could be used as a measure of the inlet pressure of the column. The two steps required for the injection of a plug could be successively executed by switching two external six-port valves (Rheodyne MX, Germany) simultaneously (Figure 3).
The separation experiments were performed using a mobile-phase composition of methanol–water (30/70, v/v). A phosphate buffer with a pH of 7 was applied for the water phase. For the separation of the three-component mixture, a solution containing of coumarin C440 (0.75 × 10–4 M), C460 (0.75 × 10–4 M) and C480 (3 × 10–4 M) (Fluka) was prepared in methanol–water (30/70, v/v). For the separation of the two-component mixture, a solution of C440 (0.75 × 10–4 M) and C460 (10–4 M) was used. Prior to use, all solutions were filtered (dpor = 0.2 μm) (Millipore, Belgium) to avoid blockage of the microchannels.
For the detection of the fluorescent sample plugs, an inverted microscope (Axiovert 200, Zeiss NV, Belgium) was used, equipped with a UV–1 filter cube set allowing for excitation at 350 nm and for emission above 450 nm. In this way, the standard mercury-vapour lamp (HBO103/W2, Zeiss) of the microscope could be used to excite the fluorescent coumarin dyes in the UV. The maximum emission of the fluorescent light occurred around 460 nm. The separations were visualized using an air-cooled CCD fluorescence camera (ORCA-ERC4742, Hamamatsu Photonics, Belgium) mounted on the video adapter of the microscope. The video images were subsequently analysed with the accompanying simple–PCI 5.1 software. The microscope was mounted on a breadboard (M–IG 23–2, Newport, The Netherlands), together with a linear displacement stage (M–TS100DC.5, Newport) and a speed controller (MM, 4006 Newport).
The operation of the pressure controllers, the external valves, as well as the translation stage was automated using a home-made software program written in C++ allowing the simultaneous control of all these instruments from one central computer.
Results and Discussion
Figure 1 shows how the Bosch process (consisting of the cyclic sequence of etching and side-wall passivation steps) allows the production of pillars with perfectly vertical side walls in silicon. The observed indentations or scallops are caused by the cyclic nature of the etching process. In each cycle, fluorocarbons (inhibiting the etching) are deposited on the bottom and side walls of the structure that is being recessed. In the subsequent etching step, these fluorocarbons are much faster removed at the bottom of the channel, while the side walls of the pillars remain much longer covered and are, therefore, attacked much less by the etching plasma, allowing them to retain a nearly perfectly vertical slope.


Figure 1
The presence of the indentations can, however, be assumed to have no significant effect on the band broadening, because the indentations only lead to a velocity inequality on the scale of approximately 0.05 μm. Given that the time needed to wipe out velocity inequalities is proportional to the square of the distances over which they need to be eliminated, the flow inequalities of the pillar indentations can be assumed to be removed (0.5 μm/0.05 μm)2 , which is 100 times faster than the velocity inequality spanning the gap between the through-pore centre and the pillar surface. Consequently, the contribution of the indentations to the total plate height can be assumed to be 100 times smaller than that of the total through-pore. The effect of the indentations on the pressure drop can be assumed to be more important. A detailed calculation showed that the indentations increase the surface with about 20% compared with a pillar with the same outer surface area but without indentations. Considering that the pressure-drop depends on the surface to liquid volume ratio according to the second power, it can be assumed that an indented pillar of the type used in the present study creates about a 1.22 , which is a 1.44 times larger pressure drop than a plain pillar. A positive aspect of the indentations is that the created surface increase can be expected to yield an enhanced retention power.
An important prerequisite for a well-performing HPLC system is its capability to withstand high pressures. Ensuring high pressure-resistant couplings has in the past proven to be a major concern for lab-on-a-chip devices.17 However, with the introduction of the commercial Nanoport-coupling systems, leakage-free connections up to 103 bar can be certified on silicon wafers. For glass chips, and using a proprietary capillary fixation technology,18 leakage-free operations up to 320 bar have already been realized. The currently used set-up was tested to pressures up to 60 bar, without observing any leakage. The weakest link of the employed set-up is probably the interface between the nano-port and the chip.
The employed design for the mobile phase distribution and sample injection channels is depicted in Figure 2. It was selected and developed to ensure a maximal uniformity of the width of the injected bands over the entire width of the channels. The channels were purposely made relatively wide (note that they have a depth over width ratio of about 100) to study a system with a reasonably high mass loadability, because the etching of pillar arrays that are deeper than 20–30 μm is not possible with the current performances of the Bosch etcher. Attempting to etch deeper pillar arrays would create flow-through pores that would be much smaller at the bottom than at the top of the pillars. This would in turn lead to an excessive band broadening.


Figure 2
To become competitive with traditional wide-bore HPLC columns in terms of mass loadability, the width of the pillar array columns will have to be of the order of a few cm or more. The problem of transiting from the traditionally employed circular connection tubing to channels with a flat-rectangular cross-section without generating too much additional band broadening should, therefore, be considered as one of the major problems in the development of pillar array columns.
In the current set-up, this problem is circumvented by performing on-chip injections as described in Figures 2–3. This also circumvented problems caused by the uneven liquid flow distribution in the bifurcating inlet system of the original Regnier design.1,13

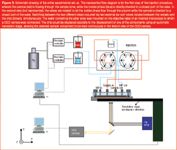
Figure 3
As can be noted from Figure 4, showing the injection and the subsequent transport of a non-retained coumarin C460 plug (the mobile phase was pure 100% methanol), the injected bands have a very rectangular shape and, for example, do not display the trapezoidal shape that is so characteristic to the bands that are obtained with electrokinetically driven injections.19


Figure 4
The reader should note that the width of the injected bands is not fixed by the width of the injection zone but can be more or less freely selected. Larger injection bands (up to 300% of the original injection band) can be formed by simply increasing the duration of the first step of the injection procedure. But also bands that are smaller than the 200 μm wide injection zone can be produced by inducing a partial leakage at port 3 during the second step of the injection procedure. With the present set-up, band widths down to a value of about of 80 μm could be produced.
Figure 4 also shows that the bands, once injected, remain perfectly straight and only broaden very slowly. In reference 14, the broadening of the unretained species bands in a channel with dp = 10 μm pillars was measured as being as low as 2 μm. In the channel shown in Figure 4, the pillars had a dp of 5 μm pillars, and the measured band broadening lead to an H value of 1 μm, leading to the same very low h value of 0.2.14
A key point in guaranteeing a reproducible and sharp injection is the complete automation of the injection set-up. Figure 5 shows four consecutive injections, all injected in a time span of only 8 seconds. The nearly indistinguishable shape of the subsequently injected tracer bands clearly demonstrates the ability of the set-up to inject in a very reproducible way.


Figure 5
The two sharp peak fronts situated near the side walls of the channel and running behind with respect to the main peak that can be observed in both Figures 4 and 5 because the flow resistance near the side walls of the channels is slightly different than in the rest of the channel. This leads to a side-wall effect that will be studied later in more detail and that has also already been partly discussed in reference 14.


Figure 6
The coating of the channels turned out to be less straightforward than anticipated. The main problems that occurred were clogging and blockage of the channels. In the instance of the 49% porosity channels, only one of the eight undertaken attempts was successful (i.e., it did not show any visible clogging particles). In the instance of the 70% porosity channels, about three of the five coated channels were clog-free. The channels that were only partially clogged still allowed the passage of a small flow. Despite the hindered flow, signs of retentive behaviour of the column could be noted. This means that in each instance the chemical bonding reaction of the C18 chains had occurred on the surface of the pillars. The partially clogged channels were, however, not useable in separation experiments because the clogging caused a significant amount of dispersion.

Figure 7
Figure 6 shows a separation for the ε = 49% channel, where the coating appeared clog-free to the eye. As can be noted from the presented compilation of subsequent video-frames, the three component coumarin dye mixture clearly separated in a distance of less than 1.5 mm and in less than 2.5 seconds. This shows that, despite the use of non-porous pillars, the retention strength of the employed pillar array column is still sufficiently strong to perform rapid separations. For the employed 70/30 (v/v) water/methanol mobile phase, the measured retention factors were k' ≈ 0, k' =1.17 and k' = 2.17 for coumarin C440, C460 and C480 respectively. The reader should note that the presented video frame images do not give an accurate image of the band broadening, because the contrast has been enhanced to yield a sufficient picture quality. Because the video frames shown in Figure 6 do not allow for an accurate determination of the band broadening, we also recorded time domain chromatograms by reading out the pixel intensity averaged over a single row of pixels running perpendicular to the flow direction and covering only the inner 50% of the channel width. Figure 7 shows the obtained response recorded at a distance of 1.25 mm downstream from the injection zone. From this response, the plate heights of the three were calculated using the peak width at half height w1/2:


after subtraction of the contribution of the width of the injected band, which can clearly not be neglected under the present condition.
The motivation to consider only the inner 50% of the channel was our desire to obtain information about the intrinsic band broadening of the pillar arrays, devoid of the side-wall band broadening effects already discussed in reference 14 and also found to be of a major influence in the present study. The plate height values obtained for the three peaks shown in Figure 7 were H = 0.9 μm, H = 2.7 μm and H = 4.9 μm for coumarin C440, C460 and C480 respectively, for a mobile phase velocity of 1.04 mm/s.
These values are clearly smaller than can be expected in a packed bed filled with particles with the same diameter (i.e., 5 μm) as the micropillars investigated here. Obviously, a part of this gain is because the non-porous pillars are being used, but as explained in, for example, references 6 and 20, the contribution of the porous zone band broadening for velocities around the minimum of the Van Deemter curve is marginal compared with the contributions coming from the mobile zone. Consequently, no major difference can be expected between the minimal plate height of a bed packed with porous compared with one packed with non-porous particles. This was, for example, recently confirmed in reference 21. Considering that the Cs-term constant in packed column is typically not larger than 0.02,22 and considering that the optimal reduced velocity in a packed column is of the order of ν = 3,8,15 it can be easily verified that stationary zone band broadening makes up only a very small fraction (hc = 0.02 × 3 = 0.06) of the typically expected total reduced plate height of h = 2. The small plate height values recorded in the present study (all smaller than h = 1) can, therefore, be fully attributed to the increased order and the consequently strongly reduced (or even completely eliminated) eddy-dispersion band broadening.
A less favourable trend in the measured plate heights is that the band broadening increases very rapidly with the retention factor, much more strongly than in a porous column. Whether this is a result of a fundamental property of ordered pillar array columns or not needs to be further investigated.


Figure 8
Figure 8 shows a separation of coumarin C440 and C460 in one of the channels where ε = 70%. Here the bands remain straight over the entire width of the channel, except (again) in the two side-wall regions. This channel was one of the few coated channels we could produce where the C18-coating was sufficiently uniform. Considering the plate heights that can be calculated from the chromatogram recorded at 1.20 mm downstream of the injection point (Figure 9), equal to H = 2.4 μm and H = 7.1 μm for coumarin C440 and C460 respectively, it can be concluded that they are significantly larger than in channel where ε = 49%. This strong increase is not unexpected. Going from the ε = 49% to the ε = 70% channel, this size increases with a factor of about 2.4 (measuring the pore necks from the SEMs yielded a value of dpor = 1.48 μm versus dpor = 3.54 μm). Since the band broadening in the pillar array can be assumed to be totally controlled by the through-pore size, this large increase in pore size obviously allows the large plate height increase observed by going from the ε = 49% channel to the ε = 70% channel to be better understood.
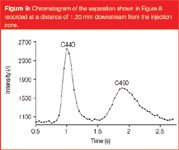
Figure 9
Conclusions
In this article we have overcome a number of practical hurdles related to the external tubing connection, flow reproducibility, the requirement of nanolitre injection volumes, and the need for supply channels distributing the sample and the mobile phase flow evenly over the pillar array to perform the first pressure-driven separations in a photolithographically-etched micropillar array column.
The observed plate heights varied between 1 (non-retained component) and 5 μm (retained component with k' = 2.17). In reduced plate heights this corresponds to h values of 0.2 and 1 respectively. The separation time needed for a three-component coumarin mixture was less than 2.5 s. The present article shows that micromachining not only helps to overcome the packing problem of capillary columns, but also leads to performances that are totally unachievable with sphere packings.
The need to establish a procedure that reproducibly yields a uniform pillar surface coating is the main problem that needs to be overcome before this novel type of column technology can be developed into a practical and commercially useful instrument. Another future research goal is to find methods to transfer from non-porous to fully meso-porous pillars.
Acknowledgments
The authors acknowledge a research grant (SBO) from the Instituut voor Wetenschap en Technologie (IWT, Belgium). H. Eghbali, is supported through a grant from the Flemish Fund for Scientific Research of the Belgian Government (FWO-Vlaanderen). D. Clicq is a Postdoctoral Fellows of the same institute (FWO-Vlaanderen).
Hamed Eghbali is a first year PhD student at the transport modelling and analytical separation science group in the department of Chemical Engineering of the Vrije Universiteit Brussel, Belgium. His position is funded through a pre-doctoral grant of the Flemish Fund for Scientific Research of the Belgian Government (FWO-Vlaanderen). His research focuses on the development of novel chip-based chromatographic devices.
Wim De Malsche is a third year PhD student in both the mesoscale chemical systems group at the University of Twente, Enschede, The Netherlands, and the transport modelling and analytical separation science group of the department of Chemical Engineering of the Vrije Universiteit Brussel, Belgium. His research focuses on the use of state-of-the-art micromachining techniques to develop novel chip-based chromatographic devices.
David Clicq has a PhD in Applied Sciences since 2004 and works at the transport modelling and analytical separation science group in the department of Chemical Engineering of the Vrije Universiteit Brussel, Belgium. He is a post-doctoral research fellow of the Flemish Fund for Scientific Research of the Belgian Government (FWO-Vlaanderen). His research focuses on the development of novel chip-based chromatographic devices.
Han Gardeniers is a professor in Chemical Engineering, and leads a research chair on mesoscale chemical systems at the University of Twente, Enschede, The Netherlands. His research focuses on the development of microfabricated chemical synthesis and analysis systems and on the fundamental aspects of reacting fluids in confined environments.
Gert Desmet is professor in Biochemical Engineering and Analytical Biochemistry from the Vrije Universiteit Brussel, Brussels, Belgium. He is leading the transport modelling and analytical separation science group in the department of Chemical Engineering of the same university. His research focuses on the miniaturization of separation methods and on the investigation of flow effects in chromatographic systems.
References
1 B. He et al., Anal. Chem., 70(18), 3790 (1998).
2 J. Kirkland and J.J. DeStefano, J. Chromatogr. A, 1126(1–2), 50–57 (2006).
3 P. Gzil et al., Anal. Chem., 76(22); 6707–6718 (2004).
4 J. De Smet et al., Anal. Chem., 76(13), 3716–3726 (2004).
5. J. Billen et al., J. Chromatogr. A, 1073(1–2), 53–61 (2005).
6. J.H. Knox, J. Chromatogr. A, 960(1–2), 7–18 (2002).
7. J.H. Knox, J. Chromatogr. A, 831(1), 3–15 (1999).
8. H. Poppe, J. Chromatogr. A, 948(1–2), 3–17 (2002).
9. F.E. Regnier, J. High Resol. Chromatogr., 23(3), 19 (2000).
10. B.E. Slentz et al., J. Sep. Sci., 25(15–17), 1011 (2002).
11. W.J. Park et al., Surface and Coating Technology, 171(1–3) 290 (2003).
12. M. Madou, Fundamentals of Microfabrication, CRC Press, London (1997).
13. K.B. Mogensen et al., Electrophoresis, 25(21–22), 3788–3795 (2004).
14. M. De Pra et al., Anal.Chem., 78(18), 6519 (2006).
15. H. Poppe, In "Chromatography, fundamentals and applicatons of chromatograph and related differential migratin methods", E. Heftmann, Ed. (Journal of Chromatography Library, Elsevier, Amsterdam, The Netherlands, 1992) pp. 151–225.
16. E. Chmela et al., Anal. Chem., 74(14), 3470–3475 (2002).
17. G. Ocvirk et al., Anal. Meth. Instrum., 2(2), 74–82, (1995).
18. R.E. Oosterbroek et al., Microsyst. Technol., 12(5), 450–454 (2006).
19. S.C. Jacobson, S.V. Ermakov and J.M. Ramsey, Anal. Chem., 71(15), 3273–3276 (1999).
20. U.D. Neue, HPLC Columns: Theory, Technology, and Practice (John Wiley & Sons, New York, USA, 1997).
21. N. Wu, Y. Liu and M. L. Lee, J. Chromatogr. A, 1131(1–2), 142–150 (2006).
22. J. Knox and H.P. Scott, J. Chromatogr. A, 282, 297 (1983).
















Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.





Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.

