Article
LCGC Europe
LCGC Europe
Practical Comparison of Different Approaches to Speed Up LC Separations
Author(s):
This article describes and compares a number of approaches to increase the speed of liquid chromatographic separations. On a standard LC column, a gain of a factor two in the run time (from 10 to 5 minutes) was achieved by increasing the flow-rate two-fold. On a monolithic column, a column operated at high temperature (120 ?C) and a short column, flow-rates could be increased typically five-fold, resulting in run times in the order of 2 minutes. This was accompanied by a sometimes considerable loss in separation efficiency. A combination of a very short run time and unaffected separation efficiency was realized on a UPLC system, designed for use at higher pressure. By working at approximately 800 bar, the analytes could be well separated within 30 seconds.
The ever-increasing need for rapid-turnaround sample analysis has led to a growing interest for fast separations, particularly in the field of liquid chromatography (LC).1 To increase the speed of analysis, a number of approaches can be followed. Very generally, the LC column dead time t0 can be taken as a representative for the overall speed of analysis and because:


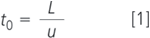
where L and u are the column length and linear mobile phase velocity, respectively, analyses can be accelerated by using shorter columns and/or higher mobile phase flow-rates. Adapting these parameters, however, affects N, the number of theoretical plates, a measure of the column efficiency:

Here, H is the height equivalent to a theoretical plate; it is roughly proportional to the particle size of the stationary phase, dp, and its dependency on u can be described by the Knox equation:2
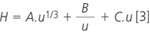
with A, B and C being system parameters representing the quality of a particular column. The reduction of the column length to speed-up the analysis will, therefore, always result in a proportional decrease in plate number and thus reduce separation efficiency. The influence of the mobile phase flow-rate on the plate height depends on the particle size (Figure 1). In general, increasing the flow-rate above its optimum level will lead to an increase in plate height and, consequently, to a decrease in separation efficiency, but this effect is much less pronounced for columns packed with small particles. A good way to improve the speed of analysis without unduly affecting the separation efficiency is, therefore, to increase the mobile phase flow-rate through a column packed with small particles. A technical limitation of this approach is the fact that the system backpressure ΔP will easily increase to a level that cannot be handled by conventional analytical instrumentation, because:
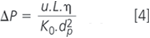
where η is the viscosity of the mobile phase and K0 the permeability coefficient of the column.

Figure 1
To overcome the pressure limitations when working with small particles and/or high flow-rates, in recent years a number of new LC stationary phases have been developed. In one approach, the permeability of the stationary phase has been considerably increased. It consists of a single rod of silica- or polymer-based material and is, therefore, called a monolithic LC phase. It contains relatively large through-pores (typically 2 μm) that are responsible for a high permeability (typically three–fold higher than in comparable packed-bed columns) and smaller mesopores (12 nm), which ensure sufficient surface area for uncompromised retentivity.3–5 In this way, analytical run times can be reduced at least two- to three-fold by simply increasing the mobile phase flow-rate.6–8
Another way to reduce system backpressure is decreasing the mobile phase viscosity. For a given mobile phase composition, this parameter decreases with increasing temperature, typically by a factor of two to three when going from 25 °C to 100 °C.9 Traditionally, the approach of high-temperature LC has always been limited by the fact that conventional silica-based LC phases are stable only up to 60 °C under reversed-phase conditions, as the underlying silica support is readily hydrolysed at higher temperatures.
However, improved column packings are now available, based on either polydentate silica, which is stable up to 100 °C, or on non-silica supports, such as carbon or zirconia, which can be used even up to 200 °C.10–13 Together with especially designed high-temperature LC hardware for mobile phase preheating, these stationary phases allow the application of two- to three-fold higher mobile phase velocities, resulting in a proportional decrease in run time, without exceeding the pressure limits of normal analytical equipment.14–17
Alternatively, high backpressures can be avoided by simply reducing the column length. Although a shorter column will lead to a decrease of the plate number, by optimizing the values for column length, particle size and flow-rate, a workable situation may be found, in which a short run time is combined with an acceptable separation efficiency and backpressure. This approach is extensively used in contemporary LC, particularly by using fast gradients, and can lead to very short run times.1,18–22
The limitations of traditional LC systems with regard to handling increased backpressures have been overcome with the recent introduction of specialized instrumentation that can operate routinely up to ca. 1000 bar.23 This instrumentation is referred to as ultra performance LC (UPLC). UPLC was designed to use fully porous 1.7 μm particle packed columns and operate them at a high linear velocity in a low dispersion environment (< 100 μL system volume) that is capable of withstanding high backpressures, so that theoretical efficiency may be reached. These attributes, combined with high-speed detection modules, allow for dramatic improvements in speed while maintaining separation efficiency and resolution.
In the work presented here, a practical comparison was made of the various approaches to improve speed in LC. Using five test compounds, the cardiovascular drug digoxin, three of its metabolites and an internal standard, the performance of a standard LC column, a monolithic column, a short column, a column at high temperature and a UPLC column was assessed in terms of analytical run time, system backpressure and plate number. In all instances, the shortest possible run time was determined at which baseline resolution (RS ≥ 1.5) between the analytes could still be achieved without exceeding the pressure limit, which (except for UPLC) was arbitrarily set at 150 bar as a typical routine high-pressure limit. A discussion of advantages and disadvantages of the different systems for use in analysis in a routine environment is provided.
Experimental
An overview of the various LC columns used is presented in Table 1. Conventional LC equipment was used in all experiments, with the exception of the high-pressure work, where the Acquity UPLC instrumentation (Waters, Milford, Massachusetts, USA) was used. In the experiments at high temperature, an in-house designed heating system with independent temperature control for eluent preheating, column heating and eluent cooling was used24 and a 500 psi backpressure regulator (GammaAnalysenTechnik, Bremen, Germany) was placed behind the detector to keep the mobile phase in the liquid state.


Table 1: Overview of LC columns.
In all experiments, linear gradient elution was performed using a mobile phase consisting of 0.2% formic acid in water and 0.2% formic acid in acetonitrile. For the conventional column, a gradient was applied of between 10% and 50% acetonitrile; for the monolithic column, the short column and the column operated at high temperature, the gradient was between 20% and 50% of acetonitrile and for the UPLC column it was between 20% and 70% acetonitrile. Mobile phase flow-rates were varied for all columns.
Column temperatures of 40–60 °C were used, except for the high-temperature experiments, which were performed between 60 and 120 °C. All the experiments used a 15 μL injection of the five-analyte mixture at a concentration of 5 μg/mL. The sample diluent was mobile phase A (with the lowest modifier concentration of the particular gradient), UV detection was performed at 220 nm.


Table 2: Results for the standard column (150 à 4.6 mm, 3.5 μm particles).
All LC phases were evaluated in terms of speed, separation efficiency and backpressure. As a measure of the speed, the time after which the last analyte had eluted was taken; re-equilibration times were not included in the evaluation. To assess the separation efficiency, separate isocratic runs were performed with digoxigenin to establish the column plate number. In all instances, the highest backpressure over the chromatographic run was recorded.

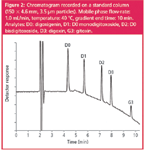
Figure 2
Results and Discussion
Standard column: As a typical conventional approach for separating five analytes, a 150 × 4.6 mm column, packed with spherical 3.5 μm particles, used at a temperature of 40 °C and a mobile phase flow-rate of 1.0 mL/min, was selected to serve as a reference. The chromatogram presented in Figure 2 shows that under these conditions the analytes are very well separated within 10 minutes, the resolution between D2 and D3, the most closely eluting compounds, being 3.8. To speed up the analysis on this standard column, the flow-rate was increased. The results summarized in Table 2 and the chromatogram in Figure 3 show that the run time can very simply be reduced two-fold by increasing the flow-rate from 1.0–2.0 mL/min.
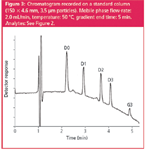
Figure 3
The accompanying increase in backpressure can be kept below the acceptable level by running at an elevated temperature of 50 °C. Although the plate number drops considerably (by 50%), it is still more than sufficient for resolving the analytes (resolution between D2 and D3: 2.7). Part of the explanation for the reduced plate number at higher flow-rates has to be related to the fact that the column plate height increases at higher velocities, (see Figure 1).
In addition, there is probably also a significant contribution of extra-column band-broadening, for example, caused by the limited acquisition speed of the detector. As this was a practical rather than a theoretical study, it was not attempted to further optimize the plate number and the obtained results have to be taken as representative for a routine analytical environment.
Altogether, the results illustrate that, without any dramatic adaptations of the chromatographic system and even without changing the column, a substantial gain in speed can be obtained on a standard LC column. The principle behind this is in fact that an excess in separation efficiency can be turned into speed by simply increasing the flow-rate until either backpressure or resolution becomes limiting. Increasing the column temperature to up to 60 °C, at which standard silica-based packings are still stable, can be helpful to control backpressure.


Table 3: Results for the monolithic column (100 Ã 4.6 mm).
Monolithic column: Silica-based monolithic columns are currently available with lengths between 25 and 100 mm and an i.d. of 4.6 mm. For the present study, a 100 mm C18 monolith was selected and chromatograms were recorded with flow-rates increasing from 1.0 to 5.0 mL/min (results in Table 3). At the conventional flow-rate of 1.0 mL/min, the performance of the monolithic column was comparable with the standard column in terms of run time, but at a more than two-fold reduced backpressure. The plate number was 30% lower than that of the standard column, which correlates to its reduced column length. Just as for the standard column, increasing the flow-rate leads to shorter run times, but because of the much lower permeability, the mobile phase can be delivered at a speed of up to 5.0 mL/min before the pressure limit is exceeded. As a consequence, the run time can be reduced five-fold to just 2 minutes. Again, the plate number decreases with increasing flow-rate, but for the present application this is not to an unacceptable level. As is illustrated in Figure 4, the resolution between D2 and D3 (1.7) is still sufficient for a baseline separation.

Figure 4
Overall, the separation performance of the monolithic column tested here is equivalent to that of a packed column of the same length. Its major advantage is that run times can be reduced to a typically two- to three-fold higher extent, albeit at the further expense of separation efficiency. A very attractive aspect of the use of monolithic LC columns is the fact that they do not need special chromatographic hardware for their operation; all it takes is simply replacing a standard packed-bed column and increasing the flow-rate. A drawback is that, up to now, monolithic columns are only available with a conventional diameter of 4.6 mm. This means that they need rather high volumetric flow-rates to speed up the analysis, which limits their compatibility with mass spectrometric detection. The availability of narrow-bore (for example, 2.0 mm) monolithic columns, which need much lower volumetric flow-rates, would, therefore, be an enormous step ahead.

Table 4: Results for the short column (20 à 2 mm, 2.5 μm particles).
Short column: To study the applicability of short LC columns for speeding up analytical run times, a column 20 mm in length was selected. As shown in Table 4, the flow-rate on this narrow-bore column can be increased up to 1.0 mL/min before the pressure limit is exceeded and this leads to a run time of just 1.8 min (Figure 5). In fact, the linear flow-rates tested on this short column are about similar to those tested on the monolithic column and also the results for run time and backpressure are comparable.

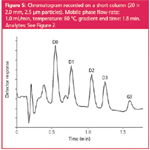
Figure 5
This means that reducing the column length and decreasing column permeability (by using a monolith) are equally useful for speeding up analytical runs. The major difference is the much lower plate number. This is, in part, because of the short bed length of the column, but again system volume and detector acquisition speed have to play an important role. Nevertheless, baseline separation (RS = 1.7) between D2 and D3 was still achieved at a run time of just 1.8 min and no further optimization was attempted.

Table 5: Results for the column at high temperature (125 à 2 mm, 5-μm particles).
An advantage of the short column approach is the availability of narrow-bore columns and, thus, the possibility to apply volumetric flow-rates compatible with MS detection. Although plate numbers are generally low, the use of relatively small particles enables the application of high flow-rates without a dramatic (further) loss in separation efficiency. Since LC–MS applications often do not need very high plate numbers, because of the high selectivity of the detection, short, narrow-bore columns, packed with small particles and operated at high linear flow-rates are an attractive means of reducing analytical run times. An additional benefit is the fact that the columns can be used with standard chromatographic equipment.
Column at high temperature: The potential of high-temperature LC was investigated using a silica-based column material, which was found to be stable up to at least 120 °C.25 Table 5 shows the results for the maximum flow-rates that could be applied at the different temperatures. By increasing the temperature up to 120 °C, the flow-rate can be increased up to 1.3 mL/min without exceeding 150 bar. In terms of linear flow-rates, this is somewhat higher than obtained with either the monolithic or the short column; the analysis time was roughly equal at 1.9 min (Figure 6).

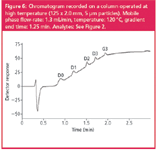
Figure 6
The lower column efficiencies are partly a result of the larger particle size, but may also be related to an inherent problem of high-temperature LC: the potential on-column degradation of the analytes. Although many compounds will be sufficiently stable during the relatively short run times at an elevated temperature, for the test compounds used here, it is known that they rapidly degrade when heated, especially at low pH.26
Another aspect worth considering is that for temperatures above 100 °C, there is a lack of stable stationary phases exhibiting the same selectivity as conventional silica-based C18 phases; in addition, the elution characteristics of the mobile phase solvents change with increasing temperature. On the one hand this might complicate transfer of existing methods, but on the other hand, it also offers additional possibilities to tune the selectivity of a chromatographic separation.
UPLC column: UPLC demonstrates the possibility of combining a very short run time and essentially unaffected separation efficiency (Table 6). When compared with the standard column, the column length is three-fold shorter and a five-fold higher linear flow-rate can be applied, which explains the considerable gain in speed. This improvement in speed is conducted while separation efficiency is kept comparable with that obtained on the standard column by the use of very small particles. Not less importantly, the results also illustrate the significance of optimized chromatographic hardware: the UPLC system was specifically designed to minimize extra-column band broadening both in the LC and optical detector modules, with a total system volume below 100 μL. In addition, the fast acquisition speed of the detector is essential to quantify these low volume chromatographic peaks.


Table 6: Results for the UPLC column (50 à 2.1 mm, 1.7 μm particles).
Altogether, the five analytes are well separated (the resolution between D2 and D3 being 3.5 and between D0 and D1 2.8) in a run time of only 30 seconds (Figure 7). This means that the run time is improved 20-fold compared with the standard column, which is about four times faster than the minimum run time of any of the other systems.

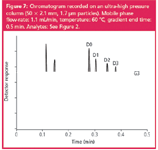
Figure 7
In all other approaches to reduce run times, speed and/or separation efficiency are sacrificed to be able to keep the backpressure at a level that can be handled by standard analytical equipment. By applying UPLC, this is not necessary. As an additional advantage, the volumetric flow-rates necessary on this narrow-bore column are comparable to those typically applied on a conventional column (in the order of 1 mL/min), which enables an easy coupling to MS detection.
Conclusions
The results presented in this paper show that chromatographic run times can very often be reduced significantly if conventional LC conditions are adjusted. For any application, running below maximum backpressure or above the required minimum resolution means that there is room for improvement. In such a situation, the analysis can be accelerated by increasing the mobile phase flow-rate until pressure or resolution becomes limiting.
A gain of a factor 2 should often be possible on conventional columns, while an additional factor 2 to 3 can be gained if a monolithic column, a short column or a column at high temperature is applied. Monolithic columns seem to be especially advantageous for conventional HPLC with its need for relatively high plate numbers and its ability to handle high volumetric flow-rates, while short columns appear to be more suited for LC–MS, which relies on the selectivity of the detector rather than chromatographic resolution.
When using specifically designed equipment that can cope with higher pressures (up to ca. 1000 bar), the run times can be further reduced: when compared with a conventional LC system, a reduction in analysis time by a factor of 20 should be possible, without compromising separation efficiency. An important general observation is that extra-column parameters such as LC system volume and detector acquisition rate are of increasing importance when speeding up the analysis.
Acknowledgement
The authors express their thanks to Eric S. Grumbach (Waters Corporation, Milford, Massachusetts, USA) for the UPLC work and valuable discussions during the preparation of the manuscript.
References
1. P.R. Tiller, L.A. Romanyshyn and U.D. Neue, Anal. Bioanal. Chem., 377, 788–802 (2003).
2. C.F. Poole, The essence of chromatography, Elsevier, Amsterdam, 2002.
3. N. Tanaka et al., Anal. Chem., 73, 421A–429A (2001).
4. N. Tanaka et al., J. Chromatogr. A, 965, 35–49 (2002).
5. K. Cabrera, J. Sep. Sci., 27, 843–851 (2004).
6. Y. Hsieh et al., Rapid. Commun. Mass Spectrom., 16, 944–950 (2002).
7. N.C. van de Merbel and H. Poelman, J. Pharm. Biomed. Anal., 33, 495–504 (2003).
8. N. Barbarin et al., J. Chromatogr. B, 783, 73–83 (2003).
9. H. Chen and Cs. Horvath, Anal. Meth. Instrum., 1, 213 (1993).
10. C. McNeff et al., LCGC, 18(5), 514–529 (2000).
11. C.J. Dunlap et al., Anal. Chem., 73, 598A–607A (2001).
12. J. Nawrocki et al., J. Chromatogr. A, 1028, 1–30 (2004).
13. J. Nawrocki et al., J. Chromatogr.A, 1028, 31–62 (2004).
14. B. Yan et al., Anal. Chem., 72, 1253–1262 (2000).
15. T. Greibrokk and Th. Andersen, J. Sep. Sci., 24, 899–909 (2001).
16. J.D. Thompson and P.W. Carr, Anal. Chem., 74, 1017–1023 (2002).
17. P. He and Y. Yang, J. Chromatogr. A, 989, 55–63 (2003).
18. L. Romanyshyn and P.R. Tiller, J. Chromatogr. A, 928, 41–51 (2001).
19. J. Ayrton et al., J. Chromatogr. B, 709, 243–254 (1998).
20. A.P. Watt et al., Anal. Chem., 72, 979–984 (2000).
21. C. De Nardi et al., J. Chromatogr. B., 762, 193–201 (2001).
22. Y. Hsieh et al., Rapid Commun. Mass Spectrom., 15, 2481–2487 (2001).
23. M.E. Swartz, J. Liq. Chrom., 28, 1253–1263 (2005).
24. T. Teutenberg et al., J. Chromatogr. A, 1114, 89–96 (2006).
25. D. Guillarme, S. Heinisch and J.L. Rocca, J. Chromatogr. A, 1052, 39–51 (2004).
26. T. Sonobe, S. Hasumi and T. Yoshino, J. Pharm. Sci., 69, 410–413 (1980).
Nico van de Merbel is scientific director at the bioanalytical laboratory of PRA International. His field of work includes method development for pharmacokinetic and pharmacodynamic assessments using chromatographic techniques. Thorsten Teutenberg is a scientist working at the Institute of Energy and Environmental Technology. His main research field is the development of innovative chromatographic methods and techniques, including high-temperature liquid chromatography. Sascha Giegold is a PhD student at the same department working on the improvement of efficiency of chromatographic separations by applying high-temperature liquid chromatography.







Newsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.

: An HPLC 2025 Video Interview with Torgny Fornstedt")