Practical Applications of Charged Surface Hybrid (CSH) Technology
Special Issues
Despite recent advances in column technology and stationary phase chemistry, challenges still remain for routine method development and LC/MS applications.
APPLICATION BENEFITS
- Wide selectivity range for method development
- Superior peak shape for basic compounds due to improved loading capacity in low-ionic strength mobile phases commonly used for LC/MS (i.e., formic acid and acetic acid)
- Ideal for pH switching applications
WATERS SOLUTIONS
ACQUITY UPLC® CSH™ and XSelect™ HPLC columns
ACQUITY UPLC and ACQUITY UPLC H-Class systems
KEY WORDS
Method development, LC/MS, ACQUITY UPLC CSH, XSelect, selectivity, pH switching, superficially porous particles
INTRODUCTION
Despite recent advances in column technology and stationary phase chemistry, challenges still remain for routine method development and LC/MS applications. This application note discusses how Charged Surface Hybrid (CSH) Technology provides solutions to the major problems still encountered with many commercially-available stationary phases, while providing a wide selectivity range for method development and general purpose use.
The reproducible addition of a low-level positive charge allows ACQUITY UPLC CSH and XSelect HPLC columns to provide different selectivity and improved performance under challenging chromatographic conditions. As a result, these columns give superior peak shape for basic compounds due to improved loading capacity under low-ionic strength conditions, as well as rapid re-equilibration after changing mobile phase pH. The practical use of columns built on CSH Technology will be demonstrated using commercial drug substances under routine use conditions. The advantages of using these columns for impurity profiling in stability indicating methods and/or forced degradation studies will be demonstrated using typical LC/MS conditions.
EXPERIMENTAL
Sample
Acid/base/neutral (ABN) mix: flavone (30 μg/mL), octanophenone (100 μg/mL), imipramine (100 μg/mL), amitriptyline (100 μg/mL), 1-pyrenesulfonic acid (50 μg/mL), diclofenac (100 μg/mL), and fenoprofen (100 μg/mL) in 75:25 H2O/CH3OH
pH switching mix: 300 μg/mL metoprolol and 100 μg/mL of amitriptyline in 95/5 H2O/CH3CN.
Base/neutral (BN) mix: Mixtures containing 2 to 200 μg/mL each of quetiapine and propiophenone were prepared in 50:50 CH3CN/H2O
Impurity mix: 500 μg/mL of imipramine spiked with 0.5 μg/mL of amitriptyline (0.1% impurity). Prepared in H2O.
METHOD
LC conditions
All LC conditions are specified in the figure captions.
MS conditions
MS system: SQ Detector
Ionization mode: ES+
Acquisition range: m/z 100 to 600
Capillary voltage: 3 kV
Cone voltage: 30 V
Data management:
Empower™ 2 CDS
RESULTS AND DISCUSSION
Selectivity
Resolution of any chromatographic separation depends on the efficiency, retentitvity, and selectivity of the column and conditions being used. Efficiency can be increased mechanically by using smaller particle columns and instrumentation with lower dispersion. Retentivity and selectivity can be altered chemically through the use of different base particles, ligand coverage, and ligand type for the packing material. Therefore, during method development experiments, exploring a variety of stationary phases with different selectivity provides the best chance to separate compounds in a mixture. However, quantitating selectivity changes across a range of stationary phases presents a challenge due to the high dependency on the conditions and analytes being used.
In order to quantitate selectivity differences, we employed a previously described method1 that compares the square of the correlation coefficient (R2) to calculate the selectivity difference (S-value) between two sets of conditions (i.e., two columns, high and low pH mobile phase, organic solvent, etc.). The S-value will be 0 for two conditions that give identical selectivity, and 100 for completely orthogonal selectivity.
This method was used to compare the selectivity of ACQUITY UPLC CSH Columns under the same set of conditions with a diverse set of compounds (Figure 1). The ACQUITY UPLC BEH C18 Column was included in the comparison as a benchmark. The calculated S-values are shown to illustrate the differences in selectivity between the four stationary phases.



Figure 1. Analysis of the ABN mix on ACQUITY UPLC CSH and ACQUITY UPLC BEH columns under low pH conditions. The ACQUITY UPLC System was equipped with a PDA detector and four-position column manager. Mobile phase A was 0.1% formic acid in water, and mobile phase B was 100% acetonitrile. The gradient was from 5 to 95% B in 5 minutes. Flow rate was 0.5 mL/min. UV detection at 254 nm. Injection volume was 2 μL. Column temperature was 30 °C. Column dimensions were 2.1 x 50 mm, 1.7 μm.
The first observation is that the application of a low-level surface charge on the CSH C18 column results in selectivity differences compared to the BEH C18 column for the ionized analytes. Secondly, the three ACQUITY UPLC CSH Columns have very different selectivity from each other, which makes them ideal choices for incorporation into a column selection protocol. Finally, this low-level charge vastly improves the peak shape for the basic compounds in low-ionic strength mobile phases, which will be discussed in more detail in the following sections.
Chromatographic selectivity can also be manipulated through the use of different mobile phase pH and elution solvent. This further enhances the capability of the columns to resolve multiple peaks in a sample. Finding stationary phases that are chemically stable above a mobile phase pH of 8 is challenging due to the pH limitation for high purity silica phases.
Both the ACQUITY UPLC CSH C18 and ACQUITY UPLC CSH Phenyl-Hexyl columns are stable at elevated pH (up to pH 11) and can thus be used with a wide range of separation conditions to yield dramatically different selectivity (Figure 2). Changing the pH of the mobile phase affects only the ionizable compounds, while the neutral compounds do not change retention time. Changing the elution solvent from acetonitrile to methanol results in longer elution times for all compounds as expected, and can change the selectivity of the separation, as is shown in Figure 2 at high pH.


Figure 2. Analysis of the ABN mix on ACQUITY UPLC CSH C18 using different mobile phase pH and elution solvent. The ACQUITY UPLC System was equipped with a PDA detector and four-position column manager. The low pH mobile phase was 0.1% formic acid in water, and the high pH mobile phase was 0.1% ammonium hydroxide in water. Mobile phase B was either 100% acetonitrile or 100% methanol. The gradient was from 5 to 95% B in 5 minutes. Flow rate was 0.5 mL/min. UV detection at 254 nm. Injection volume was 2 μL. Column temperature was 30 °C. Column dimensions were 2.1 x 50 mm, 1.7 μm.
Performance in low-ionic strength mobile phases
Method development protocols that rely on using the same columns at both high and low pH must be flushed and re-equilibrated prior to use between different pH conditions. With MS-compatible mobile phases such as formic acid and acetic acid, changes in both retention and selectivity are observed for ionized analytes2,3 at low pH after the same column has been exposed to high pH. This is likely due to a slow surface equilibration on high purity packing materials, which essentially have very little surface charge at low pH. Thus, changes in the surface charge (e.g., caused by switching from low to high pH and back again) can cause drastic shifts in the chromatographic performance, especially in low-ionic strength mobile phases.
Since ACQUITY UPLC CSH and XSelect HPLC columns already have a fixed low-level charge on the surface, re-equilibration of the stationary phase is very rapid, and no changes in retention and selectivity are observed when switching between low and high pH mobile phases on the same column (Figure 3). On other materials, such as the hybrid coated silica column shown in Figure 3, there is not only a shift in retention time for ionized bases, but also a loss in peak shape, which affects the ability to reproducibly integrate these compounds.

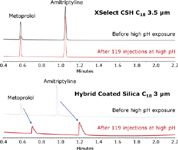
Figure 3. Analysis of the pH switching mix on XSelect CSH C18 and a silica C18 column with a hybrid coating in formic acid conditions before and after exposure of each column to 119 injections (~6 hours) at high pH. The ACQUITY UPLC System was equipped with a PDA detector and four-position column manager. The low pH mobile phase was 0.1% formic acid in water, and the high pH mobile phase was 10 mM ammonium bicarbonate, pH 10. Mobile phase B was 100% acetonitrile. The gradient was from 5 to 95% B in 2.5 minutes. Flow rate was 0.8 mL/min. UV detection at 260 nm. Injection volume was 2 μL. Column temperature was 30 °C. Column dimensions were 2.1 x 50 mm.
This degradation in performance is not due to a deterioration of the stationary phase or loss of bonded phase, since the retention and peak shape of neutral analytes are not affected.4 It is due to the fact that many commercially-available, high-purity stationary phases take an extremely long time to re-equilibrate in low-ionic strength mobile phases like formic acid after exposure to higher pH mobile phases (pH ≥7).
Another advantage of CSH columns is the ability to load larger amounts of basic compounds without deterioration in peak shape, which has many advantages for analytical and preparative development.
Many modern stationary phases suffer from poor chromatographic performance for basic compounds at low pH.5 The performance is even worse for bases analyzed using low-ionic strength mobile phases such as formic acid. While the exact cause for this behavior is not completely known, it is thought that variations in the surface charge of high purity chromatographic materials can have a large impact on the chromatography, so much so that efficiency and peak capacity for bases at low pH can decrease by more than 50%, even at fairly low analytical loads.
In order to compare the loadability of different stationary phases at low pH, a mixture of a neutral and a basic compound were analyzed at different mass loads on an ACQUITY UPLC CSH C18 Column, a fully porous silica C18 column, and a superficially porous C18 column using formic acid mobile phases (Figure 4). As the mass load increases for the neutral compound (propiophenone), all three columns exhibit symmetrical peak shape and no mass overloading is observed.

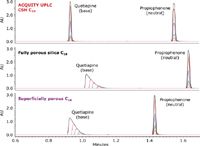
Figure 4. Loading capacity for quetiapine (base) and propiophenone (neutral) on three different columns in formic acid conditions (BN mix). The ACQUITY UPLC System was equipped with a PDA detector. Mobile phase A was 0.1% formic acid in water, and mobile phase B was 100% acetonitrile. The gradient was from 5 to 95% B in 2.5 minutes. Flow rate was 0.6 mL/min. UV detection at 254 nm. Injection volume was 5 μL. Column temperature was 30 °C. Column dimensions were 2.1 x 50 mm, 1.7 μm. The mass load on-column was between 10 ng and 1 μg for each compound.
However, when the mass load of quetiapine (a weak base) is increased on the three columns, differences in performance can be observed. The fully porous silica C18 and superficially porous C18 columns quickly become overloaded under the same gradient conditions for this compound. This occurs with a mass load as low as 50 ng on-column (20 μg/mL). The phenomenon is even worse for highly basic compounds such as amitriptyline and nortriptyline (data not shown). Conversely, the ACQUITY UPLC CSH C18 Column did not show mass overload for any of the concentrations of quetiapine, indicating that the performance of this column is identical for all analytes including basic compounds, especially in low-ionic strength conditions.
The ability to load large amounts of basic compounds in low-ionic strength mobile phases has many practical implications. These types of mobile phases are preferred for use in LC/MS applications, since there is little charge competition between components usually found in buffers, which are known to suppress the signal in mass spectrometry. This is the ideal mobile phase system for impurity profiling and identifying unknown impurities in stability indicating methods and/or forced degradation studies. However, one must inject a large amount of the active pharmaceutical ingredient (API) in order to see low-level impurities in the baseline.

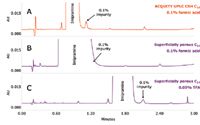
Figure 5. UV chromatograms for the analysis of the impurity mix on ACQUITY UPLC CSH C18 and superficially porous C18 columns. The ACQUITY UPLC H-Class System was equipped with a PDA detector. An SQ Detector was used for mass spectrometry analysis. Mobile phase A was water, mobile phase B was 100% acetonitrile, mobile phase C was 2% formic acid in water, and mobile phase D was 2% TFA in water. The gradient was from 25 to 35% B in 2 minutes, followed by an increase to 95% B in 1 min. Different percentages of mobile phase C and D were programmed to get the concentration of acidic modifier shown. Flow rate was 0.6 mL/min. UV detection at 254 nm. Injection volume was 5 μL. Column temperature was 40 °C. Column dimensions were 2.1 x 50 mm, 1.7 μm.
To demonstrate this, a 0.5 mg/mL solution of imipramine spiked with 0.1% of amitriptyline was injected onto ACQUITY UPLC CSH C18 and superficially porous C18 columns under formic acid conditions (Figure 5). Since amitriptyline elutes on the tail end of the imipramine peak, this example represents the challenges faced by LC/MS scientists who are routinely trying to identify unknown low-level impurities in a new synthesis pathway or forced degradation sample. Since imipramine is massively overloaded on the superficially porous C18 column, the ability to see the 0.1% impurity on the tail is lost in formic acid conditions (Figure 5B). Resolution can be improved on this column by substituting formic acid with TFA (Figure 5C). However, when trying to get mass spectral information on unknown impurities in the baseline, TFA is not a desirable mobile phase additive, since it severely suppresses ionization. In the case of the superficially porous C18 column, the impurity peak cannot be seen in the total ion chromatogram, and thus, no molecular weight information can be obtained (Figure 6B). Since the imipramine peak is not heavily overloaded on the ACQUITY UPLC CSH C18 Column under formic acid conditions (Figure 5A), one can obtain both full resolution of the impurity and molecular weight information in the same run (Figure 6A).

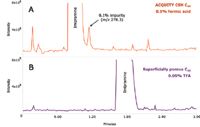
Figure 6. Total ion chromatograms for the analysis of the impurity mix on ACQUITY UPLC CSH C18 and superficially porous C18 columns. All conditions are identical to Figure 5.
CONCLUSIONS
- ACQUITY UPLC CSH and XSelect HPLC columns give a wide range of selectivity for ionizable compounds, making them ideal for inclusion in a column selection protocol and/or routine method development.
- Selectivity can be manipulated with mobile phase pH and different elution solvents. Since two of the three CSH columns are stable at high pH, they can be used under a wide variety of conditions to achieve the desired separation.
- The presence of a reproducible, low-level, fixed charge on the surface of ACQUITY UPLC CSH and XSelect HPLC columns allows for their rapid equilibration in pH switching protocols. This makes them ideal for use in method development where the mobile phase pH is frequently changed.
- ACQUITY UPLC CSH and XSelect HPLC columns have excellent loadability for basic compounds, and can be routinely used in LC/MS applications with low-ionic strength mobile phases (e.g., formic acid).
References
1. Neue UD, O'Gara JE, Mendez, A. Selectivity in reversed-phase separations: Influence of the stationary phase. J. Chromatogr. A 2006, 1127, 161.
2. Marchand DH, Williams LA, Dolan JW, Snyder LR. Slow equilibration of reversed-phase columns for the separation of ionized solutes. J. Chromatogr. A 2003, 1015, 53.
3. McCalley DV. Overload for Ionized Solutes in Reversed-Phase High-Performance Liquid Chromatography. Anal. Chem. 2006, 78, 2532.
4. Iraneta PC, Wyndham KD, McCabe DR, Walter TH. A review of Waters hybrid particle technology. Part 3. Charged surface hybrid (CSH) technology and its use in liquid chromatography. Waters white paper 720003469EN. 2010 June.
5. McCalley, DV. Study of overloading of basic drugs and peptides in reversed-phase high-performance liquid chromatography using pH adjustment of weak acid mobile phases suitable for mass spectrometry. J. Chromatogr. A 2005, 1075, 57.
Waters Corporation
34 Maple Street
Milford, MA 01757 U.S.A.
T: 1 508 478 2000
F: 1 508 872 1990













Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



