Popular Cell Expression Systems in the Biotechnology Industry
LCGC North America


The authors describe the most common cell-based protein expression systems and purification strategies used in the biotechnology industry.
Proteins are essential parts of organisms and participate in virtually every process within cells. Proteins carry multiple functions that are important in cell adhesion, signaling, immune responses, and cell cycle. Not only do they catalyze biochemical reactions, but they are also vital to cell metabolism. Therefore, the study of protein structure and function has become one of the key focuses of current research. Furthermore, many proteins possess a high intrinsic potency and selectivity for biological targets, and therefore, are very attractive as a source of therapeutic development. In the past, the manufacture of therapeutic proteins was done primarily using regular biological methods, including natural source extraction from human, animal and plant materials, nonrecombinant microbial fermentation, and regular nonrecombinant cell culture. Due to rapid technological developments and improvements in safety, the utilization of recombinant technology in high-throughput protein production is increasing. To be able to study a protein, it needs to be expressed in sufficient quantities, and for many studies, it is important to obtain a functional protein. The challenge of producing high-quality proteins at the scale required for therapeutic and commercial applications has led to the development of a variety of methods for heterologous protein production. In this column, we give an overview of the most commonly used expression systems and provide some insight on emerging expression platforms.
There are four main cell-based expression systems that are well established and widely used: bacterial, yeast, insect, and mammalian (Table I). While bacterial and yeast systems are easier to handle and scale-up, they may not deliver proteins with appropriate biological modifications, particularly for proteins of mammalian origin. Conversely, expression in insect and mammalian systems enables one to produce proteins with the most relevant posttranslational modifications (PTMs) that occur in human and other mammalian cells. Such proteins are often used as biopharmaceutical targets. It is believed that genomes of higher organisms encode for more PTMs in order to expand their structural and functional diversity (Figure 1). Bacteria are unable to perform most PTMs of eukaryotic proteins due to the lack of appropriate machinery. Yeasts demonstrate better PTMs, particularly in terms of initial stages of glycosylation, such as mannose glycosylation. However, in mammalian cells, glycosylation undergoes further processing and includes removal of mannose residues and incorporation of fucose, galactose, N-acetylneuraminic acid, N-acetylglucosamine, and other components (Figure 1). Consequently, understanding the differences and benefits of each expression system is important in deciding which system meets the essential requirements of a particular project.
Expression systems are comprised of three primary components: an expression vector, its cloned DNA containing a gene of interest (GOI), and a host for the vector that enables transfer of a foreign gene into a host cell to produce proteins. Examples of the core expression elements required in the various host-based systems are shown in Figure 2. The main goal is to express protein at a very high level, which often is referred to as overexpression. The most popular heterologous expression system is E. coli; however, the system has many limitations, one of which is expressing proteins of eukaryotic origin. Even though recent developments in protein expression and purification methodologies have led to improvements in producing soluble proteins in bacteria, these approaches are not universal and require optimization on a case-by-case basis (1,2). Not unexpectedly, a poll of our customers indicated that 80% of scientists using host-dependent protein expression systems for protein expression use multiple expression systems (Figure 3), which is consistent with current market research (3). This result is driven by challenges in the ability to express soluble and functional proteins and indicates the need to perform multisystem, low-scale optimization before large-scale protein production. A continuous search for a universal cost-effective expression system for expression of soluble proteins has led to the development of several cell-free systems that are gaining in popularity (4,5).


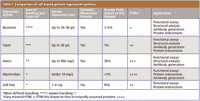
Table I: Comparison of cell-based protein expression systems
Bacterial Expression
Over the past four decades, E. coli has been the primary host for protein expression. Such broad use of bacterial systems comes primarily from the low cost of implementation in a laboratory setting, minimal technical requirements, and the very short doubling time of bacterial culture. At the same time, bacterial cultures can be easily scaled-up to incorporate the automation required for high-throughput processing. There are many examples of successful adaptations of bacterial expression systems to large-scale projects for production, including the screening of hundreds of expression clones and large-scale production of selected proteins (6–10).

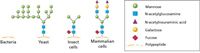
Figure 1: Typical glycosylation patterns in various cell-based expression systems. Schematic shows the different glycosylation patterns among the major protein expression systems available. While bacteria lack typical N-glycan groups, the remaining share a similar basic structure. The trimming and extension of the original N-glycan groups in mammalian cells does not occur to the same extent in yeast or insect cells.
Some other common hosts of bacterial origin are Bacillus subtilis and Brevibacillus choshinensis. For example, the Brevibacillus system has been most successfully used for assembling and expressing secreted (11) and cytoplasmic eukaryotic proteins (12–14). It produces intracellular proteins in a soluble form in the cytoplasm without forming inclusion bodies. In several cases, the Brevibacillus system works better than E. coli for expression. Therefore, B. choshinensis can be used as a host in the production of heterologous proteins irrespective of origin or nature.

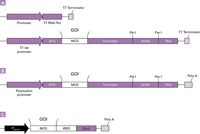
Figure 2: Examples of essential components of vector constructs for different expression systems: (a) bacterial, (b) insect, (c) mammalian. A typical gene expression cassette consists of the following major elements: A promoter, responsible for starting and ensuring the synthesis of an RNA-template encoding the gene of interest (GOI). In addition, insect and mammalian gene expression cassettes require a polyadenylation signal (poly[A]) to confer stability to the RNA-template.
Even though the bacterial expression system has been the "workhorse" in protein expression studies, this system has many limitations when it comes to expression of eukaryotic proteins. E. coli, being a prokaryote, has a very different cell structure than eukaryotes and its simpler machinery is unable to fold foreign proteins correctly and perform posttranslational modifications that commonly occur in naturally expressed eukaryotic proteins (15,16). Because eukaryotic proteins expressed by E. coli have very minimal PTMs, these proteins often are not properly folded and, therefore, are sometimes insoluble. In some cases, these limitations can be circumvented by the use of genetically modified bacterial expression strains, changing the protein purification tag or performing an additional step for protein refolding.

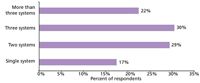
Figure 3: Examples of essential components of vector constructs for different expression systems: (a) bacterial, (b) insect, (c) mammalian. A typical gene expression cassette consists of the following major elements: A promoter, responsible for starting and ensuring the synthesis of an RNA-template encoding the gene of interest (GOI). In addition, insect and mammalian gene expression cassettes require a polyadenylation signal (poly[A]) to confer stability to the RNA-template.
Yeast Expression
Yeast cells are eukaryotic, therefore, they share the complex biology of multicellular organisms and are able to produce proteins that carry out PTMs such as proteolytic processing and lipidation. Similar to bacteria, yeast are easy to manipulate, genetically, have a rapid growth rate, and achieve high cell densities in culture, delivering up to several milligrams of protein (17–19). Most often, yeast protein expression is used for studying protein–protein interactions and protein–DNA interactions. However, recovering high amounts of desired protein from yeast cultures can be difficult due to rapid degradation of expressed proteins by endo- and exoproteases present in yeast. Also, many yeast-produced recombinant proteins are not suitable for pharmaceutical applications (20,21) due to the differences in oligosaccharide structure compared to human glycoproteins and glycosylation patterns (hypermannonization) (Figure 1).
Widely used yeasts for the production of functional proteins are Pichia pastoris, Saccharomices cerevisiae, Schizosaccharomyces pombe, and Hansanuela polymorpha. For example, yeast expression systems are used commonly for the production of G protein–coupled receptors (GPCRs), a large protein family of transmembrane receptors that sense molecules outside the cell and activate cellular responses (22). Protease-deficient P. pastoris is becoming one of the most popular yeast expression systems and has been used successfully to produce membrane proteins in amounts up to 0.35 mg/L of culture (23). Recent studies identified new transcriptional factors in S. pombe similar to those in higher eukaryotes. This opens new opportunities for utilization of S. pombe in future studies of protein function (24).
Expression in Insect Cells
Because a substantial part of protein research is focused upon mammalian and human proteins, the mammalian system is the most desirable system for many research and production projects. This system delivers fully functional proteins with the most relevant biological modifications. However, mammalian expression systems typically produce very low protein output, and require laborious scale-up processes. Consequently, it can be the least economical system among those mentioned here.
Insect cell culture addresses the need for glycosylation and three-dimensional folding among human-derived proteins, requiring these features for biological activity, while at the same time avoiding the susceptibility of mammalian cells to viral contamination that could cause health problems to humans. Proteins are expressed in insect cells such as Lepidoptera, the insect genus that includes butterflies and moths. Other commonly used insect cell types include Spodoptera frugiperda (fall army worm), Bombyx mori (silkworm), Trichoplusia ni (cabbage looper), and Malacosoma disstria (forest tent caterpillar). All the insects belonging to this group are susceptible to infection by baculovirus, and the genetic material of this insect virus serves as the vector for inserting foreign proteins into the insect cells.
Most of the baculoviral expression vectors are based upon Autographa californica multiple nuclear polyhedrosis virus (AcMNPV). The AcMNPV baculoviral particle carries 120–150 kb (kilobases) of double-stranded genomic DNA and can accommodate an additional 100 kb of foreign genetic material, which presents practically limitless capacity for insertion of single or multiple gene expression constructs. One of the most commonly used baculoviral promoters is the polyhedron promoter that regulates expression of baculoviral polyhedron protein during the late phase of the baculovirus life cycle. Polyhedron protein often can comprise up to 50% of the total cellular protein. Moreover, polyhedrin is dispensable for virus replication in cell culture. Therefore, it is usually deleted from a baculoviral expression vector and a GOI is inserted under the control of a polyhedron promoter, allowing production of up to 1 g/L of the protein of interest (POI), with an average production range of 10–100 mg/L. Protein expression in insect cells allows protein PTM (signal peptide cleavage, phosphorylation, lipid modification, and glycosylation) as well as folding (disulfide bond formation, oligomerization), similar to that in mammalian cells. Insect cell cultures (SF9, SF21, and others) are easy to scale-up and can be maintained both as adherent and suspension cells at 27 °C, which is essential for expression of temperature-sensitive mutants. As an additional advantage, the polyhedron promoter is active during the late phase of the baculovirus replicative cycle, and allows expression of toxic genes with minimal deleterious effects to the cell. Baculoviruses infect only insects and are nonpathogenic to humans. Thus, baculovirus-based protein expression requires only Biosafety Level 1 practices.
Baculovirus systems offer a solution for obtaining sufficient quantities of proteins with PTMs, similar to those obtained by mammalian systems, but with less effort. Because of its many advantages, in the past 10 years, the baculovirus protein expression system has become widely used for enzyme manufacturing, large-scale sample preparation for structural studies, and also in vaccine development (25–29).
Mammalian Expression
As described previously, recombinant eukaryotic protein expression systems have been developed in both yeast and insect systems; however, these systems still differ in PTMs as compared to those produced in mammalian cells. These PTMs can include, but are not limited to, glycosylation, sumoylation, phosphorylation, methylation, and acetylation (30). These PTMs are important because they directly relate to the functionality of the recombinant protein being produced. Therefore, mammalian protein expression systems were developed to overcome the limitations of yeast and insect systems. Typically, mammalian systems consist of a cell line that has been adapted to suspension culture in order to facilitate scale-up into bioreactor-based cultures and growth at very high densities. Typical rodent and human cell lines used for these purposes include CHO, NIH3T3, BHK, HepG2, and HEK 293. Yields from highly expressing CHO cell lines under optimum culture conditions have reached as high as 5 g/L (31). Additionally, these cultures are grown in serum-free media to reduce contamination by serum proteins and to facilitate easier purification of the protein product and faster downstream processing.
To express the POI, one must introduce a deoxyribonucleic acid (DNA) expression construct that contains not only the sequence of the protein to be expressed, but also important cis-acting expression control elements. Cis-acting regulatory elements are regions of DNA or RNA that regulate the expression of genes located on that same molecule of DNA, minimally consisting of a promoter for RNA polymerase II driven transcription and a polyadenylation signal for mRNA termination. An important factor in the overall success of the expression process is the choice of promoter that is used to drive expression of the protein. In many cases, the cytomegalovirus (CMV) immediate early promoter is used to drive high levels of constitutive expression in the chosen cell line. This constant expression, however, can be detrimental due to potential toxic or cytostatic properties of the protein being expressed. In this situation, application of an inducible expression system, such as the tetracycline inducible promoter system, might be advisable. Using an inducible system such as this permits expression only when it is desired. The tetracycline inducible system utilizes the Tet repressor protein, fused with a transcriptional activation domain, to bind to Tet operator sequences and either negatively or positively regulate the expression of your POI (32,33). Tet-inducible activation (rather than repression) of a minimal promoter allows more tunable regulation and higher fold-induction (ratio of minimal to basal expression levels) of protein expression. In fact, it has been shown that the level of Tet-inducible gene expression is higher than that from a constitutively active promoter (34).
One limitation of using a mammalian expression system for protein production is the work required to deliver the expression construct into the cell. An expression vector can be delivered into the cell as a DNA molecule with the help of a transfection reagent, allowing DNA transport through the cellular and nuclear membranes. Gene delivery and expression by transfection can be transient or stable. Stable gene expression often requires selection of cell clones, carrying an integrated vector with a positive selection marker and also expressing the POI. However, selection of transfected, high-expressing, positive cell clones requires much time and effort. A more efficient method for gene delivery to cells is infection with attenuated (nonpathogenic) viruses, converted into gene vehicles, carrying genetically engineered gene expression cassettes. The advantage of viral gene delivery is that viruses have the ability to infect almost every cell of a given cell population, thereby ensuring that the majority of cells will express the desired protein, often negating the need for screening cell clones. Viruses employed in this fashion include lentivirus (35) and adenovirus (36). An example of the production of a lentivirus vector is illustrated in Figure 4. These viral systems can provide a fast and efficient means to produce cell populations, expressing recombinant proteins in reasonable amounts with desirable PTMs.

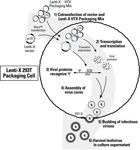
Figure 4: Overview of lentivirus production. Initially, cotransfection of a Lenti-X vector and the Lenti-X HTX Packaging Mix (Step 1) results in the production of the corresponding recombinant lentiviral genomic RNA transcript and viral packaging proteins (Step 2). Recognition of the packaging sequence (Ψ) on the recombinant viral RNA genome by the packaging proteins (Step 3) results in the assembly of viral cores, which are transported to the cell membrane (Step 4). Cores are then enveloped by cellular membrane containing aggregated VSV-G envelope proteins. Mature, infectious virions then bud from the cell (Step 5) and are collected in the medium (Step 6).
Cell-Free Expression
Protein expression also can be done in vitro (Latin: within the glass) using purified RNA polymerase, ribosomes, transfer RNA (tRNA), and ribonucleotides. These reagents can be produced by extraction from cells or from a cell-based expression system. Because protein synthesis occurs in cell lysates rather than within cultured cells, the method is also called cell-free protein expression. Cell-free systems utilize cell lysates from a variety of sources for biosynthesis of proteins by adding exogenous genetic information (37,38). The use of cell-free systems is becoming an attractive alternative to cell-based protein expression, because it offers a simple, open, and flexible approach to rapid synthesis of folded proteins. The open nature of cell-free systems makes them easily modifiable, thereby allowing addition of external molecules to create favorable conditions for protein folding and activity. For example, cell-free systems can be used to incorporate non-natural or chemically modified amino acids to assist in labeling proteins for downstream applications (4,5). A number of PTMs are achievable in eukaryotic lysates: glycosylation, peptide cleavage, acetylation, phosphorylation, arginine methylation, and others (4,37,38). The ability to incorporate unnatural or chemically modified amino acids into proteins being produced has been applied to create several protein technologies based on a cell-free system: protein in situ arrays (PISA), nucleic acid programmable protein arrays (NAPPA), DNA to protein arrays (DAPA), and others (4,5,38–42). Cell-free systems are still considered a relatively new platform. Due to the low level of protein expression and high cost, cell-free systems are not as widely adopted as cell-based systems. These systems require further optimization, particularly to improve protein yield.
Protein Purification Tags and Optimization of Protein Output
After protein is expressed, it needs to be isolated for structural and functional studies. The protein of interest can be purified as a wild-type protein or as a recombinant protein. A recombinant protein is a wild type protein that has a tag attached either at the C-terminus or N-terminus. The tag gives the expressed protein affinity toward a particular ligand. The isolation and purification of a single wild-type protein can take up to six months of work, while the isolation and purification of a single tagged protein can take up to one day. Recombinant protein purification is the preferred method for isolation and purification of the POI because it is faster and yields sufficient protein for various downstream applications.
There are many tags used, and the most popular are polyhistidine (his-tag), glutathione S-transferase (GST), FLAG peptide, maltose binding protein (MBP), c-Myc, hemagglutinin (HA) peptide, Strep, and others. These tags have various characteristics that have to be taken into consideration when choosing a purification strategy (Table II). For many projects a native target protein is desired; therefore, an affinity tag has to be removed after purification. This introduces an additional purification step that can be disadvantageous for large-scale projects.

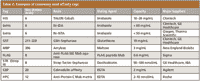
Table II: Examples of commonly used affinity tags
Immobilized metal affinity chromatography (IMAC) is one of the common techniques used for protein purification. It is based upon the reversible interaction between various amino acid side chains and immobilized metal ions (43). The most common tag used among many expression systems is a polyhistidine tag that exhibits highly selective binding to certain metals and has great utility in IMAC. Although only two to three histidines may bind transition metals under certain conditions, six histidines reliably bind transition metals in the presence of strong denaturants such as guanidinium (44). Such protein tags are commonly referred to as 6xHis tags. There are several alternative polyhistidine-purification tags, including 6xHis, 6xHN, and HAT. Histidine tags are easy to use and have become a reliable and inexpensive purification media. However, in many cases, only up to 30% of proteins are soluble when a histidine tag is attached at either end of the protein. This low solubility affects the effectiveness of the expression and purification process and therefore additional steps need to be taken to purify the protein.


Table III: Glossary
The 6xHis tag is a six-histidine peptide that binds strongly to divalent metal ions such as cobalt and nickel. 6xHis tags are small. In many cases, they do not interfere with protein expression and function, and are useful when purifying low molecular weight proteins. The 6xHN tag is a histidine-rich peptide that has similar solubility and binding characteristics to 6xHis, but is more useful than 6xHis when purifying high molecular weight proteins. Generally, it is more difficult for a resin to bind a high molecular weight protein than a low molecular weight protein because the bulk of a larger protein can interfere with the resin's ability to bind to the polyhistidine tag.


Figure 5: Choice of metal ions: specificity versus adsorption.
For IMAC protein purification, it is important to know that each metal ion has some unique characteristics. Copper has the lowest specificity and the highest adsorption for histidine-tagged proteins, and zinc is at the opposite end of the scale with higher specificity and lower adsorption (Figure 5). The most common resins for histidine-tagged protein purification are charged with cobalt or nickel. Nickel-based resins offer higher adsorption and lower specificity as compared to cobalt, which translates into a higher binding capacity for the target protein but also lower purity. Cobalt-based resins offer higher specificity and lower adsorption as compared to nickel, which translates into higher purity of the target protein and a lower capacity (45). The most popular resins for histidine-tagged protein purification are TALON and Ni-NTA. TALON has a remarkable affinity and specificity for polyhistidine-tagged proteins and does not bind native proteins that have histidine-rich sequences such as the SlyD protein expressed in E. coli (46) (Figure 6). The TALON reactive core, which contains cobalt, has strict requirements for the spatial positioning of histidines. Only adjacent histidines or specially-positioned, neighboring histidines are able to bind cobalt in this reactive core. In nickel-based resins (that is, Ni-NTA resin), these spatial requirements are less strict. Therefore, nickel-based resins are also able to bind histidines located in places other than the protein's polyhistidine-tag. Therefore, when choosing between cobalt-based resins and nickel-based resins one has to keep in mind that the tradeoff is purity versus binding capacity.


Figure 6: SlyD binds to Ni-NTA but not TALON columns. Each resin was loaded with cleared total bacterial lysates from BL21 (DE3) pLysS cells with no expressed polyhistidine-tagged protein. (a) Denaturing conditions, (b) native conditions. Lane 1: cleared lysate. Lane 2: Ni-NTA final wash. Lanes 3â5: Ni-NTA elution fractions. Lane 6: TALON Resin final wash. Lanes 7â9: TALON Resin elution.
The HAT sequence is a novel IMAC affinity tag that contains six histidines unevenly interleaved by other amino acid residues. This sequence of nonadjacent histidine residues possesses less overall charge, and a more evenly distributed charge, than tags with consecutive His residues, such as the 6xHis tag. Thus, HAT-fusion proteins have better solubility and similar affinity towards immobilized transition metal ions and zinc. In addition, HAT-fusion proteins can be adsorbed in the absence of imidazole at neutral pH. As a result, the alkaline proteases present in cell lysates are less active, and therefore most HAT-tagged proteins are more stable.
The GST-tag is commonly used as many expression vectors include a thrombin domain for cleavage of the GST-tag during purification. The GST-tag addresses the solubility issue that is common when using histidine tags. Roughly around 40% of the proteins expressed using GST-tags are soluble. The main concern with using a GST-tag is the size of the tag (up to 220 amino acids), which often interferes with protein folding and activity. There are many suppliers of gluthatione resins like GE, Pierce, Clontech, and others. Generally, the binding capacity for the glutathione resins is 10 mg/mL and there are no major differences between the resins offered by various suppliers (Table II).
While histidine-tag purification and GST-tag purification are based on affinity, epitope-tag purification is based on a tag (for example, FLAG, Myc, and HA) binding to its primary antibody. Also, some epitope tags help with expression of soluble, functional proteins. The down side is that because the purification resins are based upon a primary antibody being immobilized to beads, the yield and purity are highly dependent upon the quality of the immobilized antibody. Even though proteins eluted using epitope tags are very pure, the yields are low, therefore, this method is not cost-effective, and is only used for very specific projects.
For commercial production of protein products, other approaches are more popular. Ion exchange is the preferred chromatographic technique for the commercial recovery and purification of IgG from plasma (47). Ion exchange is also amenable to large-scale purification of monoclonal antibodies with the popularity driven by high capacities, simpler cleaning and sanitization, and robustness with respect to scale-up and large scale operations. For purification of monoclonal antibody products, Protein A chromatography serves as the capture step in the platform process (48). The Protein A step also serves as the key volume reduction step in the process since the product stream is concentrated from a relatively dilute cell culture supernatant to concentrations of >10 g/L. This step alone can result in purities of >99% due to the high selectivity.
Future Developments
As discussed earlier, the end goal of the development of these systems is to produce required amounts of properly folded, posttranslationally modified, functional protein for biochemical and biological analysis. All of the aforementioned systems have been highly optimized for ease-of-use, high yields, higher-throughput, scalability, and cost savings. Some directions for future improvements can involve addressing different aspects of the systems ranging from rational design to employment of advanced hardware. In terms of optimization of expression within mammalian and insect systems, both would benefit from the inclusion of highly evolved expression elements such as designer promoters known to express at high levels within the desired cell line. In mammalian cells, it has been observed that the activity from viral promoters, such as that from CMV, often can be silenced due to changes in chromatin status (methylation or acetylation) during extended culture. Inclusion of scaffold or matrix attachment regions (S/MAR) within the expression construct have been shown to alleviate these effects. Moreover, in the majority of cases, the expression construct is integrated randomly into the genome of the protein-producing cell, where it is subject to changes in chromatin status as mentioned previously. Recently, genome engineering has become a more attainable goal, where enzymes such as zinc-finger nucleases (ZFNs) (49) or meganucleases (50) are utilized to create double-stranded breaks in the cellular genomic DNA to facilitate homologous recombination of the expression cassette into genomic regions (safe harbors) that are known to express at high levels. Although this occurs at lower frequency than random integration, more consistent expression can be achieved. In addition to optimization of expression construct design and location, the cells themselves can be better characterized through complete analysis of transcriptional activity with the cell under various culture conditions. This permits a more rational design to cell line development and optimization of culture parameters (media, growth factors, and so forth) to identify those factors that maximize cell number and protein yield. In addition to culture components, computer-controlled feeding algorithms can be used to increase the consistency, specific-output, and yield during the growth phase of production. Taken together, these improvements can be applied to existing high-throughput methods of expression, which include rapid cloning and screening methods as well as purification on automated platforms. Cell-free advances include the application of leader sequences within the mRNA which permit more efficient translation across a number of cell extracts (51) as well as immobilization of the translation machinery to enhance output (52).
In addition, future directions in bacterial expression systems include modifications such as further inclusion of chaperones to aid in folding, codon optimization for higher translational efficiency, and elimination of secondary structure within the mRNA transcript to permit more efficient ribosome function. Currently, there is no one system that provides every protein in a high yield, functional, and scalable fashion, but with these improvements, it might become clearer which system to choose for a particular protein.
Acknowledgments
The data and figures presented in this article were obtained at Clontech Laboratories Inc, a Takara Bio company. We thank Dominique DeBold for technical review and editing of the article, Jennifer Kolanek for preparing the figures, and Ira S. Krull and Anurag S. Rathore for review and valuable suggestions regarding article content.
Thomas P. Quinn is the Leader of Viral Technologies Group at Clontech Laboratories, Inc. Tom received his Masters in Molecular Medicine and Genetics from Wayne State University (Detroit, MI). Tom has been working on the development of tools for the fields of gene therapy and viral delivery and expression systems for over 15 years and has been the key person in development of many Clontech products for retroviral, adenoviral, lentiviral and baculoviral expression systems, associated kits, and other cell biology products.
Marianne Rivkin is a Technical Support Scientist and Product Coordinator for Mammalian and Baculoviral Expression Systems at Clontech Laboratories, Inc. Marianne received her M.S. in Biochemistry and Ph.D. in Molecular Biology from Moscow State University (Moscow, Russia) and completed her Postdoctoral Fellowship at Stanford University. Marianne has over 20 years of experience in Academia and Biotech industry with particular expertise in viral research, mammalian expression, and gene therapy; she has authored over 20 publications in peer reviewed journals.
Corina R. Nikoloff is a Product Manager at Affymetrix. She received the BS degree in Microbiology and Biochemistry from California Polytechnic State University (San Luis Obispo, CA) and MBA degree from San Jose State University (San Jose, CA). Corina has extensive experience in research and development, particularly working on antibody array development and protein purification products.
Tatyana N. Kabakova is a Product Manager at Clontech Laboratories. Tatyana holds an MS degree in Biology with concentration in Microbiology from Kyiv National T. Shevchenko University (Kiev, Ukraine) and MBA degree from Golden Gate University (San Francisco, CA). Tatyana has over 10 years of experience in development and manufacturing of products for the biotech industry


Ira S. Krull Ira S. Krull "Biotechnology Today" Co-Editor Ira S. Krull is a Professor Emeritus of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.


Anurag S. Rathore Anurag S. Rathore is a biotech CMC consultant and an associate professor with the Department of Chemical Engineering, Indian Institute of Delhi, India. He is also a member of BioPharm International's editorial advisory board.
The Editors regret that they cannot accept e-mail to answer specific questions related to their columns.
References
(1) P. Braun and J. LaBaer, Trends Biotechnol. 21, 383–388 (2003).
(2) C. Langlais, B. Guilleaume, N. Wermke, T. Scheuermann, L. Ebert, J. LaBaer, and B. Korn, BMC Biotechnol. 7, 64–75 (2007).
(3) 2008/2009 North American MSPPSA Protein Expression & Purification Systems Report. (Phortech International, San Carlos, California, February 28, 2008).
(4) A. Spirin, Trends Biotechnol. 22, 538–545 (2004).
(5) A.M. Jackson, J. Boutell, N. Cooley, and M. He, Briefings Functional Genom. 2, 308–319 (2004).
(6) L. Dieckman, M. Gu, L. Stols, M.I. Donnelly, and F.R. Collart, Protein Expression Purification 25, 1–7 (2002).
(7) F. Baneyx, Current Opinion in Biotechnol. 10, 411–421 (1999).
(8) H.E. Klock, A. White, E. Koesema, and S. A. Lesley, J. Structural Functional Genom. 6, 89–94 (2005).
(9) K. Terpe, Appl. Microbiol. Biotechnol. 72, 211–222 (2006).
(10) S. Yokoyama, Current Opinion Chem. Biol. 7, 39–43 (2003).
(11) M. Mizukami, H. Hanagata, and A. Miyauchi, Current Pharm. Biotechnol. 11(3), 251–258, (2010).
(12) H. Yamagata, K. Nakahama, Y. Suzuki, A. Kakinuma, N. Tsukakoshi, and S. Udaka, Proc. Nat. Acad. Sci. USA 86, 3589–3593 (1989).
(13) Y. Takimura, M. Kato, T. Ohta, H. Yamagata, and S. Udaka, Biosci.Biotechnol. Biochem. 61(11), 1858–1861 (1997).
(14) K. Yashiro, J.W. Lowenthal, T.E. O'Neil, S. Ebisu, and H. Takagi, Protein Expression Purification 23, 113–120 (2001).
(15) M.R. Dyson, S.P. Shadbolt, K.J. Vincent, R.L. Perera, and J. McCafferty, BMC Biotechnol. 4, 32 (2004).
(16) Y.Y. Londer, S.E. Giuliani, T. Peppler, and F.R. Collart, Protein Expression Purification 62, 128–137 (2008).
(17) M.A. Romanos, C.A. Scorer, and J.J. Clare, Yeast 8, 423–488 (1992).
(18) R.M. Bill, Current Genetics 40, 157–171 (2001).
(19) S. Macauley-Patrick, M.L. Fazenda, B. Mcneil, and L.M. Harvey, Yeast 22, 249–270 (2005).
(20) C.F. Goochee, M.J. Gramer, D.C. Andersen, and J.B. Bahr, in Frontiers in Bioprocessing, Vol. 2, P. Todd, S.K. Sikdar, and M. Bier, Eds. (American Chemical Society, Washington, DC, 1992), p. 199.
(21) M.A. Romanos, C.A. Scorer, and J.J. Clare, Yeast 8, 423 (1992).
(22) H. Reilnder and H.M. Weiss, Current Opinion Biotechnol. 9, 510–517(1998).
(23) N. Andre, N. Cherouati, C. Prual, T. Steffan, G. Zeder-Lutz, T. Magnin, F. Pattus, H. Michel, R. Wagner, and C. Reinhart, Protein Sci. 15, 1115–1126 (2006).
(24) C.A.S. Banks et al., J. Biol. Chem. 282(8), 5761–5769 (2007).
(25) L. Luo and M. I. Sabara, Clin. Diagnostic Lab. Immunol. 12(8), 904–909 (2005).
(26) H. Almanza, C. Cubillos, I. Angulo, F. Mateos, J.R. Caston, W.H. M. van der Poel, J. Vinje, J. Barcena, and I. Mena, J. Clin. Microbiol. 46, 3971–3979 (2008).
(27) B. Saunier, M. Triyatni, L. Ulianich, P. Maruvada, P. Yen, and L.D. Kohn, J. Virol. 77(1), 546–559 (2003).
(28) K. Fukunaga, M. Arita, M.Takahashi, A.J. Morris, M. Pfeffer, and B. D. Levy, J. Biol. Chem. 281, 9490–9497 (2006).
(29) M.L. Heuze, F.C. Guibal, C.A. Banks, J.W. Conaway, R.C. Conaway, Y.E. Cayre, A. Benecke, and P.G. Lutz., J. Biol. Chem.. 280(7), 5468–5474 (2005).
(30) C.T. Walsh, S. Garneau-Tsodikova, and J.R. Gatto, Angewandte Chemie Int. 44(45), 7342–7372 (2005).
(31) D. L. Hacker, S. Nallet, and F. M. Wurm, BioPharm Int. Supplement (June 2, 2008), pp. 6–15.
(32) M. Gossen and H. Bujard, Proc. Nat. Acad. Sci. USA 89, 5547–5551 (1992).
(33) M. Gossen, S. Freundlieb, G. Bender, G. Muller, W. Hillen, and H. Bujard, Science 268, 1766–1769 (1995).
(34) D.X. Yin, L. Zhu, and R.T. Schimke, Anal. Biochem. 235, 195–201 (1996).
(35) B. Gaillet, R. Gilbert, S. Broussau, A. Pilotte, F. Malenfant, A. Mullick, A. Garnier, and B. Massie, Biotechnol. Bioeng. 106(2), 203–215 (June 2010).
(36) B. Gaillet, R. Gilbert, R. Amziani, C. Guilbault, C. Gadoury, A.W. Caron, A. Mullick, A. Garnier, and B. Massie, Biotechnol. Progress 23(1), 200–209 (January 1, 2007).
(37) T. Endoh, T. Kanai, Y.T. Sato, D.V. Liu, K. Yoshikawa, H. Atomi, and T. Imanaka, J. Biotechnol. 126, 186–195 (2006).
(38) S. Mikami, T. Kobayashi, S. Yokoyama, and H. Imataka, J. Biotechnol. 127, 65–78 (2006).
(39) M. He, O. Stoevesandt, E. A. Palmer, F. Khan, O. Ericsson, and M.J. Taussig, Nature Methods 5, 175–177 (2008).
(40) K.S. Anderson, N. Ramachandran, J. Wong, J.V. Raphael, E. Hainsworth, G. Demirkan, D. Cramer, D. Aronzon, F.S. Hodi, L. Harris, T. Logvinenko, and J. LaBaer, J. Proteome Res. 7, 1490–1499 (2008).
(41) A. Rolfs, W. R. Montor, S.S. Yoon, Y. Hu, B. Bhullar, F. Kelley, S. McCarron, D.A. Jepson, B. Shen, E. Taycher, S.E. Mohr, D. Zuo, J. Williamson, J. Mekalanos, and J. LaBaer, Proc. Nat. Acad. Sci. USA 105, 4364–4369 (2008).
(42) M. He, New Biotechnol. 25(2/3), 123–132 (2008).
(43) J. Porath, J. Carlsson, I. Olsson, and G. Belfrage, Nature 258, 598–599 (1975).
(44) E. Hochuli, H. Dobeli, and A. Schacher, J. Chromatogr. 411, 177–184(1987).
(45) E. Sulkowski, Bioessays 10, 170–175 (1989).
(46) J.L. McMurry and R.M. Macnab, Clontechniques XIX(1), 16–17 (January 2004).
(47) D. Low, R. O'Leary, and N.S. Pujar, J. Chromatogr., B 848(1), 48–63 (2007).
(48) A.A. Shukla, B. Hubbard, T. Tressel, S. Guhan, D. Low, J. Chromatogr., B 848(1), 28–39 (2007).
(49) S.J. Orlando, Y. Santiago, R.C. DeKelver, Y. Freyvert, E.A. Boydston, E.A. Moehle, V.M. Choi, S.M. Gopalan, J.F. Lou, J. Li, J.C. Miller, M.C. Holmes, P.D. Gregory, F.D. Urnov, and G.J. Cost, Nucleic Acids Res., epublished ahead of print (June 8, 2010).
(50) F. Paques and P. Duchateau, Current GeneTherapy 7(1), 49–66 (February 1, 2007).
(51) S. Mureev, O. Kovtun, U.T.T. Nguyen, and K. Alexandrov, Nature Biotechnol. 27(8), 747–752 (August 2009).
(52) S. Park and K. Hamad-Schifferli, ACS Nano 4(5), 2555–2560 (May 25, 2010).

















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

