Pharmaceutical Regulations: An Overview for the Analytical Chemist


The pharmaceutical industry develops and manufactures life-saving medicines and is regulated by government authorities to ensure drug products’ safety, efficacy, and quality before reaching patients. This article provides a high-level overview of pharmaceutical regulations and associated public quality standards.
The pharmaceutical industry develops new drugs to address unmet medical needs (1). Health authorities heavily regulate the industry to ensure drug product quality in late-stage development and commercial production. While the complex and multi-faceted regulatory process has been described in books, reports, and source documents (1–5), this paper provides a high-level overview of pharmaceutical regulations and their processes for the analytical chemist. The topic is particularly interesting to the separation scientist who conducts many pivotal laboratory studies in analytical and quality assessment to support drug product releases and to provide critical data for regulatory filings using lab equipment such as high performance liquid chromatography (HPLC) with ultraviolet (UV) detection (6).
This paper is the fourth in a series of nine papers on “pharmaceutical industry for the analytical chemist.”
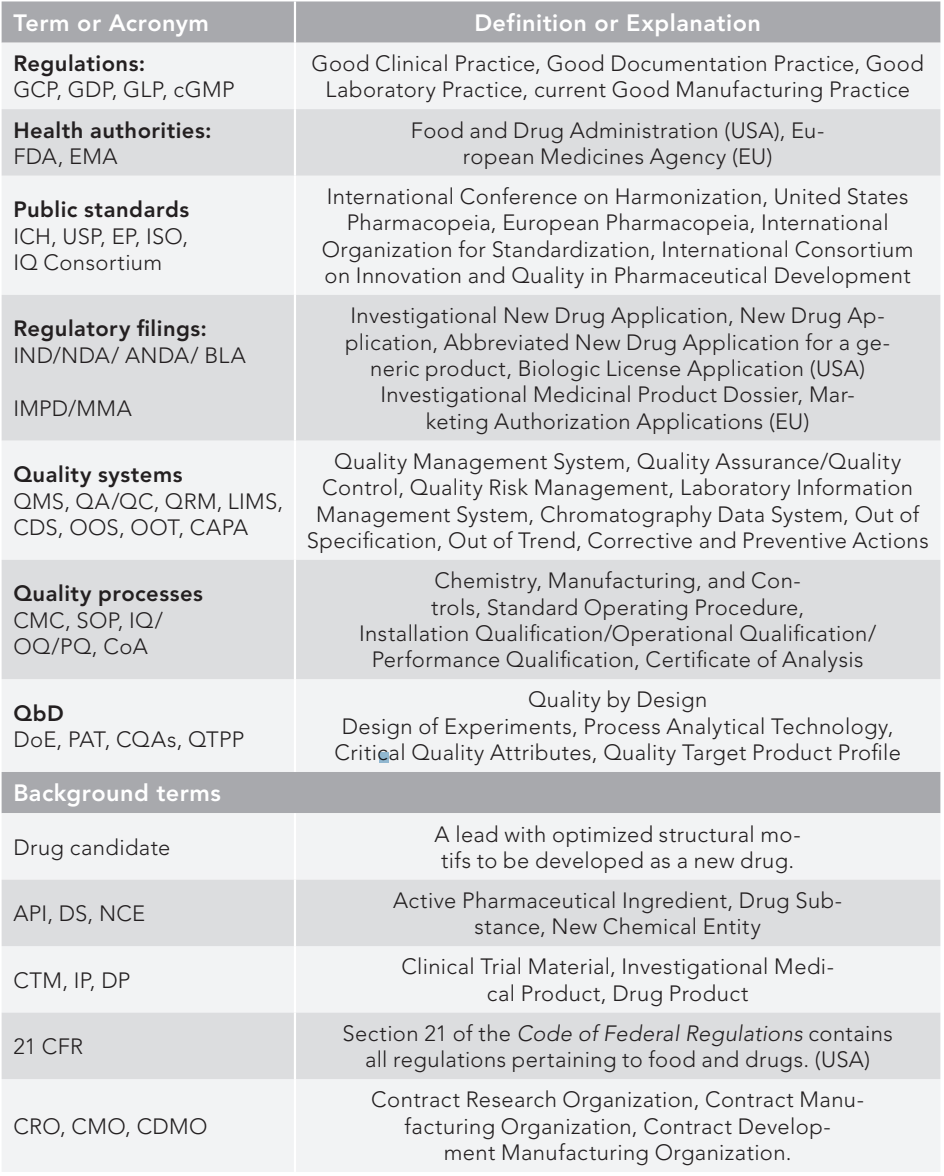
Table I lists the terms and acronyms used in the paper. Figure 1 is a pictorial portrayal of the regulatory processes categorized into five sections to be discussed in the text later: 1. Pharmaceutical regulations; 2. Health authorities; 3. Public standards; 4. Pharmaceutical organizations; and 5. Regulatory trends/approaches.


Figure 1: A pictorial portrayal of the pharmaceutical regulatory processes, categorized into five sections for discussion in the text.
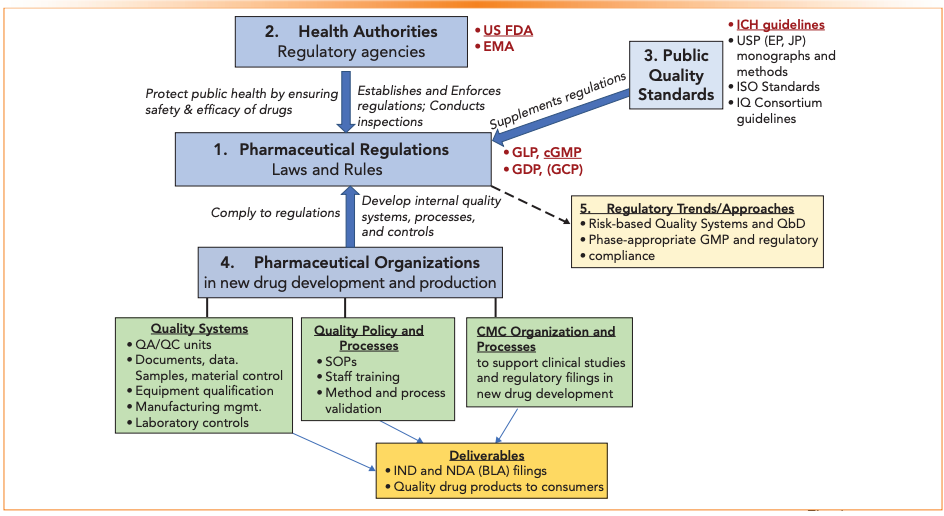

Table I: Glossary of terms and abbreviations (categorized)
Section 1: Pharmaceutical Regulations
Pharmaceutical regulations aim to ensure drug safety, efficacy, and quality. Regulations are established by government agencies to interpret laws and facilitate practical implementation. Government health authorities enforce regulations. The general laws governing pharmaceutical companies in the USA are recorded in the Code of Federal Regulations Title 21 (21CFR); how a company sets up its systems, facilities, and SOPs to meet these laws needs to be determined by each company. While the goal of regulation is straightforward, interpretation and compliance processes by pharmaceutical companies can be elaborate, diverse, and arduous (4,5).
This section briefly describes four pharmaceutical regulations (GLP, GMP, GCP, and GDP). GLP and GMP will be discussed further in papers #5 and #6 of this series. Since the CFR is not updated regularly, it has become common to put a lowercase c in front of the acronym, such as cGMP, to keep in mind that good practice must meet currently accepted standards.
Good Laboratory Practice (GLP)
The GLP regulations, described in 21 CFR 58 (7), is a system of management controls for laboratories to ensure consistency and reliability of results in nonclinical studies. GLP regulations are followed by laboratories, animal care facilities, and CROs conducting animal toxicology evaluations and bioanalytical clinical studies. GLP uses distinctively different terminologies and languages from GMP regulations, focusing on shorter-term projects and standalone studies. Laboratories in pharmaceutical manufacturing facilities follow GMP regulations in general; for more details in analytical studies, non-governmental references, such as Pharmacopeia and ICH, are followed.
Current Good Manufacturing Practice (cGMP)
cGMP is a regulation and guidance governing the manufacture and testing of API, diagnostics, foods, pharmaceutical products, and medical devices. It is described in 21 CFR 210 and 211 (8) and ICH Q7 (9) for small molecule drugs. The GMP regulations for biologics are described in 21 CFR 600 and 610, while those for medical devices and foods are described in 21 CFR 812 and 820, and 21 CFR 110, respectively.
In particular, 21 CFR 211 lists the detailed regulatory requirements for various regulatory expectations on organization, facilities, equipment, control of components/production process/packaging, distribution, laboratory testing and controls, records/ reports, and returned drug products.
Good Documentation Practice (GDP)
GDP describes standards by which critical documents are created and maintained to ensure their integrity and traceability (10). GDP standards are not codified in a single 21 CFR, but their compliance is required in GMP facilities. GDP recommends that records be completed and written according to specific standard operating procedures (SOPs) from the initial data generation to recording and processing, use, retention, archiving, and retrieval. The guidance for electronic records and signatures is covered in 21 CFR Part 11 (11). Recording of information/data during the manufacture and QC testing must be done in an Accurate, Legible, Contemporaneous, Original, and Attributable (ALCOA) process.
Good Clinical Practice (GCP)
Good Clinical Practice (GCP) is an international quality standard that governments transpose into regulations for clinical trials involving human subjects (12). There is no single regulatory document in the CFR on GCP, though the requirements are found in ICH E6 (13). The goals and principles of GCP are the assurance of ethics in clinical trials, compliance with the study protocol, informed consent and protection of confidentiality of the test subject, and quality assurance of CTM.
Section 2: Health Authorities
Governmental health authorities are regulatory agencies that establish and enforce rules, laws, and policies required to ensure consistent drug safety, efficacy, and quality. In recent years, health authorities have harmonized many expectations, but jurisdiction-specific regulations and criteria remain. Pharmaceutical companies must submit regulatory filings to a relevant health authority before conducting clinical trials (IND and IMPD) to gain approval for marketing distributions of new drugs (NDA, ANDA, MA, BLA).
US FDA and EMA
The Food and Drug Administration (FDA) is the regulatory authority in the United States over new drugs and devices (14). FDA reviews research data and information about investigational drugs (IND) and new drug applications (NDA/ANDA or BLA). They monitor drug safety/efficacy/quality issues and drug information and advertising. Each regulatory authority typically aligns with public quality standards, such as ICH and corresponding regional pharmacopeias.
A primary regulatory activity of the FDA’s Center for Drug Evaluation and Research (CDER for small molecules and CBER for biologics) is reviewing developmental drug products through clinical phases I, II, and III trials and the marketing approval. For example, nonclinical laboratory studies must follow GLP, and Phase IIb studies and beyond must comply with cGMP. These regulations are typically risk-based and phase-appropriate.
EMA, the European Medicine Agency, is the counterpart to the FDA in the European Union.
Section 3: Public Quality Standards
Augmenting the regulations are public quality standards, or guidelines to which pharmaceutical companies generally adhere, even though they are not considered regulations.
International Conference on Harmonization (ICH)
ICH brings together regulatory authorities of Europe, Japan, and the USA and pharma experts to discuss scientific and technical aspects of product registration. ICH issues guidance documents in Quality, Safety, Efficacy, and Multidisciplinary topics (QSEM) (15). For example, the overall pharmaceutical development guidelines are described in ICH Q8 and Q11, and the common technical document format (CTD) is described in ICH M4Q. For analytical chemists in pharma, ICH Q1 (stability), Q2 (validation), Q3 (impurities), Q6 (specifications), Q9 (risk management), and Q12 (life cycle management) are commonly referenced external guidance documents.
Pharmacopeial Standards
Regional compendia, such as the United States Pharmacopeia (USP) (16), British Pharmacopeia (BP), European Pharmacopeia (EP), and Japanese Pharmacopeia (JP), establish quality standards for the attributes, analytical procedures, and acceptance criteria of excipients, drug substances, dosage forms, and compounded preparations. Testing and compliance with the standards described in compendial methods are essential for manufacturing, releasing, and distributing pharmaceutical ingredients and generic drug products. These compendia are updated periodically, and one must follow the current version at the time of testing.
United States Pharmacopeia (USP)
USP is a non-governmental organization whose mission is to improve world health by setting public standards to help ensure the quality, safety, and benefits of medicines and foods (16). They issue the USP–NF, a combination of two compendia, the United States Pharmacopeia and the National Formulary.
USP monographs contain tests, procedures, and acceptance criteria. Monograph testing requires the use of compendial reference standards. The USP also contains general chapters on standard techniques such as chromatography <621>, pH <791>, dissolution <711>, content uniformity <905>, and analytical life cycle <1220>. System suitability attributes for HPLC methods, such as the number of theoretical plates, tailing factor, and resolution, are often derived from USP <621> for regulatory submissions.
The National Formulary (NF) contains monographs on excipients. A drug company wishing to formulate a drug product using an API or excipient not contained in a compendium such as NF must provide characterization, methods, validation, and stability data on the novel substance.
International Organization for Standardization (ISO)
ISO is an independent, non-governmental international organization with a membership of 167 national standards bodies (17). ISO offers certifications to a wide variety of standards. For example, pharmaceutical companies pursue ISO 9001 certification to demonstrate they have a quality management system based on seven well-established quality management principles. ISO 9001 helps ensure that the patient gets consistently high-quality products, which benefits the business.
Section 4: Pharmaceutical Organizations
Pharmaceutical regulations are specific on what is required or expected in drug products but do not generally furnish details on how to comply with these requirements. For instance, the GMP regulations state that:
“There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products….”
However, the regulations do not give details on the structure of the QC unit, the testing procedures, or the product specifications used in these approvals. Each pharmaceutical company is expected to develop its own internal organizations, systems, policies, and processes (e.g., standard operating procedures or SOPs) to ensure drug product quality (3–4). This section provides a brief overview and examples.
Internal Quality Standards, Systems, and Processes
For most pharma laboratory scientists, internal quality systems are the first examples of regulations encountered. Figure 2 provides an example of the organizational structure of quality systems and processes in a GMP manufacturing/development facility, including those within a QC laboratory.

Figure 2: An example of the organizational structure of quality systems and processes in a GMP manufacturing/development facility, including those within a QC laboratory.

Pharmaceutical companies create a Quality Management System (QMS) to align their business practices and comply with global regulations. Companies interpret and implement local policies such as SOPs or work instructions to promote traceability, quality, and compliance. Some companies have more detailed structural SOPs for internal systems, but others have a less elaborate/less structuralized approach. Audits and inspections evaluate a company’s practices against its quality systems to ensure compliance. Audit observations may lead to the issuance of a corrective and preventative actions (CAPA) investigation to resolve gaps and deficiencies. Issues found during routine QC testing can lead to CAPA investigations as well. The results of the CAPA will be a detailed plan that, when enacted, will ensure that this specific situation does not recur in the future.
Laboratory regulations such as personnel training and safety, controlled documentation (including lab notebooks), equipment qualification, electronic records/electronic signatures, reference standards programs, method validation, data review, and LIMS all serve as components covered by a QMS. There are cGMP site-level SOPs defining roles and responsibilities, quality and safety standards, analytical procedures, control of raw materials, sample handling, data reporting, CAPA systems, and change controls. Companies implement a process to investigate OOS/ OOT results using root-cause analysis. Daily operational activities such as accurate and contemporaneous record keeping; identifying reagents, balances, and equipment used; standardization; and system suitability ensure results generated by the separation scientist are accurate, traceable, and reproducible.
CMC Process in New Drug Development
While regulatory requirements of a manufacturing facility are well defined, those in the development organization of new drugs could be more interpretive, diversified, and nuanced.
CMC is an acronym for chemistry, manufacturing, and controls, encompassing critical technical activities such as formulation development, manufacturing process development, and the analytical support of the CTM for clinical trials. CMC details are required in CTD Module 2 & Module 3 for regulatory submissions for both FDA and EMA. This requires a good understanding of the physicochemical properties of the NCEs and the establishment of quality specifications designed to ensure the safety and consistency of the newly developed drug product. The typical functions and collaborative association of a “CMC” organization within a major company were described elsewhere (3).
The CMC process includes identifying planned and potential registration areas for the new drug product. This defines the regulatory authority whose applicable laws and regulations will be followed to develop a clinical trial application (IND) and, later, the new drug application (NDA or BLA).
Section 5: Strategic Approaches in Drug Development
Several recent regulatory trends and approaches are endorsed by the health authorities and captured in ICH Q7 (GMP), Q8 (R2) (Pharmaceutical Development), Q9 (Quality Risk Management), and Q10 (Pharmaceutical Quality System) (15). The underlying concepts in these approaches are discussed here, including quality by design (QbD), risk-based quality systems, and phase-appropriate regulatory compliance.
Quality by Design (QbD) and Quality Risk Management
“The quality cannot be tested into the product but should be built into it.” Joseph M. Juran (18).
QbD can be achieved with a thorough understanding of production processes during development and manufactured with a knowledge of risks and their mitigation. A systematic approach to development is encouraged by health authorities (19), which begins with predefined objectives for the intended product profile and an emphasis on understanding product, process, and process control based on science and quality risk management (QRM). The core toolsets and concepts in QbD include the Design of Experiments (DoE), QRM, Process Analytical Technologies (PAT), Quality Target Product Profile (QTPP), Critical Quality Attributes (CQAs), and the control of critical process parameters and raw materials (19).
Phase-Appropriate GMP and Regulatory Compliance
While ICH guidelines cover regulatory requirements in late-stage development, the recommended specifications may be too stringent in early development, where the characterization and processes for DS and DP are not completely defined. In this regard, guidance documents from the FDA (20) and the IQ Consortium (International Consortium on Innovation and Quality in Pharmaceutical Development) (21) may offer more reasonable recommendations in setting specifications in early-phase clinical development (before phase IIb).
Summary
Compliance with regulations described in the source documents should result in scientifically sound and traceable data which characterize a manufacturing process that consistently produce drug products with the required quality. In this concise and systematic review of the regulatory process, we hope to offer a better understanding of the underlying concepts and quality systems to the analytical chemist and how their work under a GMP environment can contribute to product quality and patient benefits.
Disclaimer
This paper provides a high-level overview of pharmaceutical regulations to the analytical chemist. The information presented here stems from health regulation documents, guidance documents, and the authors’ own experiences. Presenting a concise primer on this complex topic is challenging. We selected a simplified approach to review regulatory activities’ underlying principles and structural components with references to books, source documents, and helpful journal articles.
Acknowledgments
The authors thank the reviewers for providing timely technical and editorial inputs to this article: Alice Krumenaker of Hovione, Kate Evans of Longboard Consulting, Jenny Lin Gao from Tergus Pharma, Mike Shifflet from J&J Consumer, Lily Liu-Shin from Arcus Biosciences, and He Meng of Sanofi.
References
(1) Hill, R. G.; Rang H.P. (Eds). Drug Discovery and Development: Technology in Transition, 2nd ed., Churchill Livingston, Elsevier, 2012.
(2) Ali, J.; Baboota, S. (Eds.) Regulatory Affairs in the Pharmaceutical Industry, 1st Edition, Academic Press, 2021.
(3) Dong, M.W. HPLC and UHPLC for Practicing Scientists, 2nd Ed., Wiley, 2019, pp 221–243, 281-303.
(4) Sarker, D. K. Quality Systems and Controls for Pharmaceuticals, Wiley, 2008.
(5) Pharmaceutical Quality for the 21st Century A Risk-Based Approach Progress Report, US FDA, 2007, https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/pharmaceutical-quality-21st-century-risk-based-approach-progress-report (accessed 2023-04-18)
(6) Kou, D.; Wigman, L.; Yehl, P.; Dong, M. W. Separation Science in Drug Development, Part 4: Quality Control, LCGC North Am. 2015, 33 (12), 900-909.
(7) Code of Federal Regulations, Title 21, Part 58, Good Laboratory Practice for Nonclinical Laboratory Studies, Government Publishing Office, 2011.
(8) Code of Federal Regulations, Title 21, Parts 210 and 211, Current Good Manufacturing Practice for Finished Pharmaceuticals, Government Publishing Office, https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211 (accessed 2023-04-18)
(9) ICH Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, US FDA, 2016.
(10) Good documentation practice, Wikipedia. https://en.wikipedia.org/wiki/Good_documentation_practice (accessed 2023-04-18)
(11) Code of Federal Regulations, Title 21, Part 11, Electronic Record; Electronic Signatures – Scope and Application, US FDA, 2003.
(12) Kolman, J.; Meng, P.; Scott, G. (Ed.), Good Clinical Practice: Standard Operating Procedures for Clinical Researchers, Wiley, 2021.
(13) ICH E6 (R3) Good Clinical Practice (GCP), ICH, 2021. https://database.ich.org/sites/default/files/ICH_E6-R3_GCP-Principles_Draft_2021_0419.pdf (accessed 2023-04-18)
(14) US FDA, What We Do, https://www.fda.gov/about-fda/what-we-do (accessed 2023-04-18)
(15) ICH Guidelines, https://www.ich.org/page/ich-guidelines (accessed 2023-04-18)
(16) United States Pharmacopeia, USP, https://www.usp.org/ (accessed 2023-04-18)
(17) International Organization of Standardization (ISO), https://www.iso.org/home.html (accessed 2023-04-18)
(18) Juran, J. M. Juran on Quality by Design: The New Steps for Planning Quality into Goods and Services. Free Press, 1992.
(19) Nasr, M. M. Implementation of Quality by Design (QbD): Status, Challenges and Next Steps, CDER, FDA, 2006.
(20) cGMP for Phase 1 investigational Drugs, US FDA, 2008. https://www.fda.gov/media/70975/download (accessed 2023-04-18)
(21) IQ Consortium, https://iqconsortium.org (accessed 2023-04-18)
About the Co-Author

Leon Doneski has over 20 years of experience in the pharmaceutical industry, contributing to many product registrations. He is currently the Director of Regulatory CMC for Arcutis Biotherapeutics. Leon earned his PhD in analytical chemistry from the University of Delaware.

About the Author

Michael W. Dong is a principal of MWD Consulting, which provides training and consulting services in HPLC and UHPLC, method improvement, pharmaceutical analysis, and drug quality. He was formerly a Senior Scientist at Genentech, a Research Fellow at Purdue Pharma, and a Senior Staff Scientist at Applied Biosystems/PerkinElmer. He holds a PhD in Analytical Chemistry from City University of New York. He has more than 130 publications and a bestselling book in chromatography. He is an editorial advisory board member of LCGC North America and the Chinese American Chromatography Association. Direct correspondence to: LCGCedit@mmhgroup.com.

















Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.

Characterizing Polyamides Using Reversed-Phase Liquid Chromatography
May 5th 2025Polyamides can be difficult to characterize, despite their use in various aspects of everyday life. Vrije Universiteit Amsterdam researchers hoped to address this using a reversed-phase liquid chromatography (RPLC)-based approach.


New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

