Online Solid-Phase Extraction–Gas Chromatography–Flame Ionization Detection System for Monitoring Contaminants at Parts-Per-Trillion Concentrations in Process Waters
Online monitoring of odour and taste components that occur at parts-per-trillion (ppt) levels in industrial process waters requires specialized analytical hardware that is generally not compatible with the harsh environmental conditions in these typical industrial settings. An alternative instrumental method is proposed that uses dynamic extraction in combination with gas chromatography (GC) equipped with a simple flame ionization detector (FID) to achieve these extremely low detection limits. The extraction process was fully automated by means of online solid-phase extraction (SPE). The combination of online SPE and GC–FID was used to monitor the quality of process water contaminated with 2-methylisoborneol and geosmin, which are two notorious odour and taste components, in volumes up to 1 L.
2-Methylisoborneol (2-MIB) and geosmin (trans-1,10-dimethyl-trans-9-decalol) are two well-known off-flavours that can be produced by a wide variety of micro-organisms (1,2). They result, for example, in the typical stale odour that you come across in water that has been left unattended for a while in an uncooled plastic bottle. They are also responsible for the cork taint off-odour of wine (3,4). Because of their unpleasant odour and the fact that they are characterized by extremely low odour threshold levels, typically at the low ng/L level, both components are an important cause of product quality issues in multiple industries. Aquaculture production units and drinking water production facilities are particularly susceptible to significant production losses when either 2-MIB and/or geosmin are perceived in the final products they deliver. Therefore, these industries must regularly measure 2-MIB and geosmin concentrations to anticipate any potential quality issue well before product release. Unfortunately, accurate analysis of 2-MIB and geosmin at parts-per-trillion (ppt) concentrations—particularly in a monitoring scheme with reasonable time resolution—is far from straightforward. Obviously, sensitive and selective analytical instrumentation is mandatory, and because of the volatility of both components, gas chromatography (GC) in combination with mass spectrometry (MS) or MS/MS are the first choice. Moreover, a suitable enrichment technique is required as well, to amplify detector response well above background noise levels. Suitable techniques in that respect include headspace solid-phase microextraction (HS-SPME), which has been used quite extensively, but also stir-bar sorptive extraction (SBSE) (5–8). Both techniques are much alike and apply microextraction at equilibrium conditions. In the case of SPME, sampling is performed in headspace mode, whilst for SBSE the sampling device is submerged in the water sample and applied like a magnetic stir-bar, which improves mass transfer but complicates end‑to-end process automation. In the SPME procedure, 2-MIB and geosmin are promoted to the headspace by means of salting out, shaking, and incubating the sample at elevated temperature (40–60 °C) (7). Once in the headspace, components are trapped on the SPME fibre, which is coated with a small amount of a suitable polymeric material, such as carboxen/polydimethylsiloxane and divinylbenzene/carboxen/polydimethylsiloxane (9,10). After sampling—which can take up to 60 min for maximal sensitivity—the fibre is transferred to a hot GC inlet where the trapped components are quickly desorbed and swept to the GC column for subsequent analysis. Because of the small amount of material, these injections can be performed in splitless mode without significant peak tailing.
To prevent accidental release of 2-MIB and geosmin into the production process, online monitoring at regular time intervals would be of great industrial benefit. Unfortunately, the intended production facilities, such as water treatment plants and aquaculture production, are not properly equipped or controlled (dirty, high temperature, and relative humidity) to adequately accommodate the intended GC–MS instrumentation in close vicinity to the production line. In addition, the MS hardware, pumps, and electronics are not designed to be exposed to these harsh conditions 24/7 via continuous monitoring. In addition, the mass spectrometer requires trained operators to tune, use, and properly maintain the device. From our perspective, it would make more sense if the MS detector was replaced by a more robust and far less complicated device, such as the flame ionization detector (FID). Unfortunately, FID is less sensitive than MS, necessitating another, more effective procedure to concentrate the analytes prior to analysis. Driven by the nature of both samples and analytes, solid-phase extraction (SPE) seems to be a reasonable candidate to achieve the required degree of preconcentration, predominately because it is—contrary to SPME and SBSE—not an equilibrium technique but allows exhaustive extraction of target analytes from large sample volumes. Unsurprisingly, both 2-MIB and geosmin have been extracted successfully from aqueous samples using SPE with C18 packed cartridges (11,12). After extraction, both analytes were conveniently eluted from the cartridge by means of a suitable organic solvent, such as ethyl acetate, and the extract analyzed by GC–MS. Concentration factors of 2000 are reported in the literature (12).
An important factor that needs to be considered when applying SPE—particularly when compared with SPME and SBSE—involves the loss of sensitivity, as only a small portion of the obtained extract is injected, even when it is evaporated and/or when large-volume injection is applied. To compensate for this inherent sensitivity loss, users have only one option and that is to proportionally increase the volume of sample sent over the SPE cartridge. Unfortunately, this is not the most effective way of working, as increasing the sample volume requires the amount of packing material to be increased as well, to achieve exhaustive extraction without analyte breakthrough, which in turn increases the volume of extraction solvent for quantitative elution. To preserve the potential of SPE preconcentration, the entire extract needs to be transferred quantitatively to the GC column rather than only a portion, which is only feasible when the SPE cartridge is placed inline to the GC system during elution. Online SPE–GC is not an easy solution, mainly because of solvent incompatibility reasons when it is applied to process aqueous samples. Nonetheless, several attempts have been made, with varying success, including microextraction by packed sorbent (MEPS) and micro SPE (µSPE) in miniature cartridges (10,13). Both solutions are micro-options by design, very much aligned with common automated sample handling systems, and, therefore, direct injection of the entire elution solvent on the GC inlet is readily achievable. However, employing micro-scale configurations with minute amounts of packing material (only a few milligrams) for processing large sample volumes appears unsuited.
Initial investigation of online SPE–GC–MS for measuring pollutants in aqueous samples has been conducted by Bulterman et al. (14,15). More recently, the analysis of mineral oil hydrocarbons (MOH) in food, fats, and oils has gathered a lot of attention, mainly because of the potential risk that these components may have on public health (16). As a result, several instrument manufacturers have developed hardware solutions in which these analyses are performed using online liquid chromatography (LC)–GC–FID (16). Here, a long normal-phase silica column is used to fractionate the mineral oil saturated hydrocarbons (MOSH) from the mineral oil aromatic hydrocarbons (MOAH) and transfer both fractions to the GC system using an elegant combination of selection and backflush valves. Since both fractions are eluted with hexane and dichloromethane, respectively, quantitative transfer of both fractions to the GC system (parallel column setup, injection volumes 400 µL each) is in fact relatively easy. By the same philosophy, we developed a similar setup, replacing the LC column with a SPE cartridge, but maintaining the capability to transfer the fraction eluting from the cartridge column quantitatively to the GC system. Measures were taken to address the incompatibility between the aqueous sampling phase and subsequent elution with non-aqueous GC‑friendly solvents. In this article, we present our results on this new online SPE–GC–FID system for the preconcentration and analysis of 2-MIB and geosmin in process water.
Experimental
A short, stainless-steel cartridge (30 × 2.1 mm × ¼ outside diameter [o.d.]., Restek, Interscience) was packed with 80 mg adsorbent particles (held in place with two metal frits) and served the purpose as a SPE cartridge. The cartridge was contained within a custom valve box (Interscience), equipped with two 6-port 2-position rotary valves (VICI International) to direct sampling, purge, and elution flows. Before sampling, the bed was preconditioned by rinsing with ethyl acetate (> 99%, Sigma Aldrich) and subsequently with an excess of ultrapure water (MilliQ, Sartorius). Analyses were performed with a 25 ppt 2-MIB and geosmin solution in 1 L ultrapure water prepared from a 2-MIB and geosmin stock solution (100 µg/mL mix solution, Sigma Aldrich).
An Ultimate 3000 HPG-3200BX binary pump (Thermo Fisher Scientific, Interscience) was used to control the flow over the packed SPE bed. Pump A was used to draw the water sample and push it over the SPE bed (1 L at 50 mL/min). After sampling, the adsorbed bed was dried with nitrogen (N5.0) and the flow paths were rinsed with ultrapure water and ethyl acetate consecutively. Finally, pump B was used to deliver 150 µL ethyl acetate over the adsorbent bed (at 300 µL/min)to elute the components from the packing bed and transfer the analytes quantitatively to the GC part of the instrument.
The outlet of the SPE cartridge was connected to a Trace 1300 GC (Thermo Fisher Scientific, Interscience) by means of a Y-piece, which also served as inlet point for the GC carrier gas (helium, N5.0, supplied at 5 mL/min). The remaining outlet part of the Y-piece was connected to 15 m of a 0.53 mm internal diameter (i.d.) fused silica capillary column (uncoated, except for the last 3 m with 0.25 µm polydimethylsiloxane, Thermo Fisher Scientific, Interscience), which served as retention gap to capture the entire volume of ethyl acetate eluting from the SPE cartridge. To remove the excess ethyl acetate prior to analysis, the retention gap was not connected directly to the analytical column (15 m × 0.25 mm, 0.25-µm Rxi-5 MS Mega, Interscience), but indirectly by means of a T-piece that also included a heated solvent vapour exit (SVE) valve. Briefly, during solvent transfer—that is, adsorbent bed elution—the SVE valve was opened so that all the ethyl acetate vapours were conveniently vented away. Just before almost all the ethyl acetate was removed, the SVE valve was closed, thereby connecting the retention gap with the analytical column, and chromatography was able to start. Flow rates between 30–50 mL/min were used to evaporate the solvent. The SVE temperature was kept at 150 °C. The GC oven was operated with a ramped temperature starting at 55 °C for 4 min before ramping to a final temperature of 240 °C at 50 °C/min. The entire unit was controlled by Chromeleon CDS (Thermo Fisher Scientific, Interscience).
Online SPE–GC Instrument: An online SPE–GC–FID analyzer was developed that consists of three distinct units: the pumping unit, the trapping unit, and the analysis unit. The pumping and trapping unit are designed to process large volumes of water, preconcentrate any dissolved analytes of interest, and subsequently elute the analytes in any GC-compatible solvent. At its heart, the instrument contains a small amount of an appropriate adsorbent that is recycled continuously for maximal uptime. The trapping unit contains several switching valves to appropriately direct sample, elution, and purge flows over the cartridge. The analysis unit consists of a GC–FID system. Analyte transfer was achieved by means of a setup that is similar to large volume on-column injection.
Preconcentration Process in Pump and Trapping Unit: The adsorbent bed was used to enrich 2-MIB and geosmin from water. The minimal sample volume required was determined by the instrument detection limit (IDL). In the case of GC–FID, this implied that at least 0.1 to 1 ng of both analytes must be trapped by the adsorbent and quantitatively transferred. In the case of 2-MIB and geosmin, the desired detection limit in water was close to 1 ppt; this corresponded to sample volumes from 1 to 0.1 L (for 0.1 ng and 1 ng IDLs, respectively).


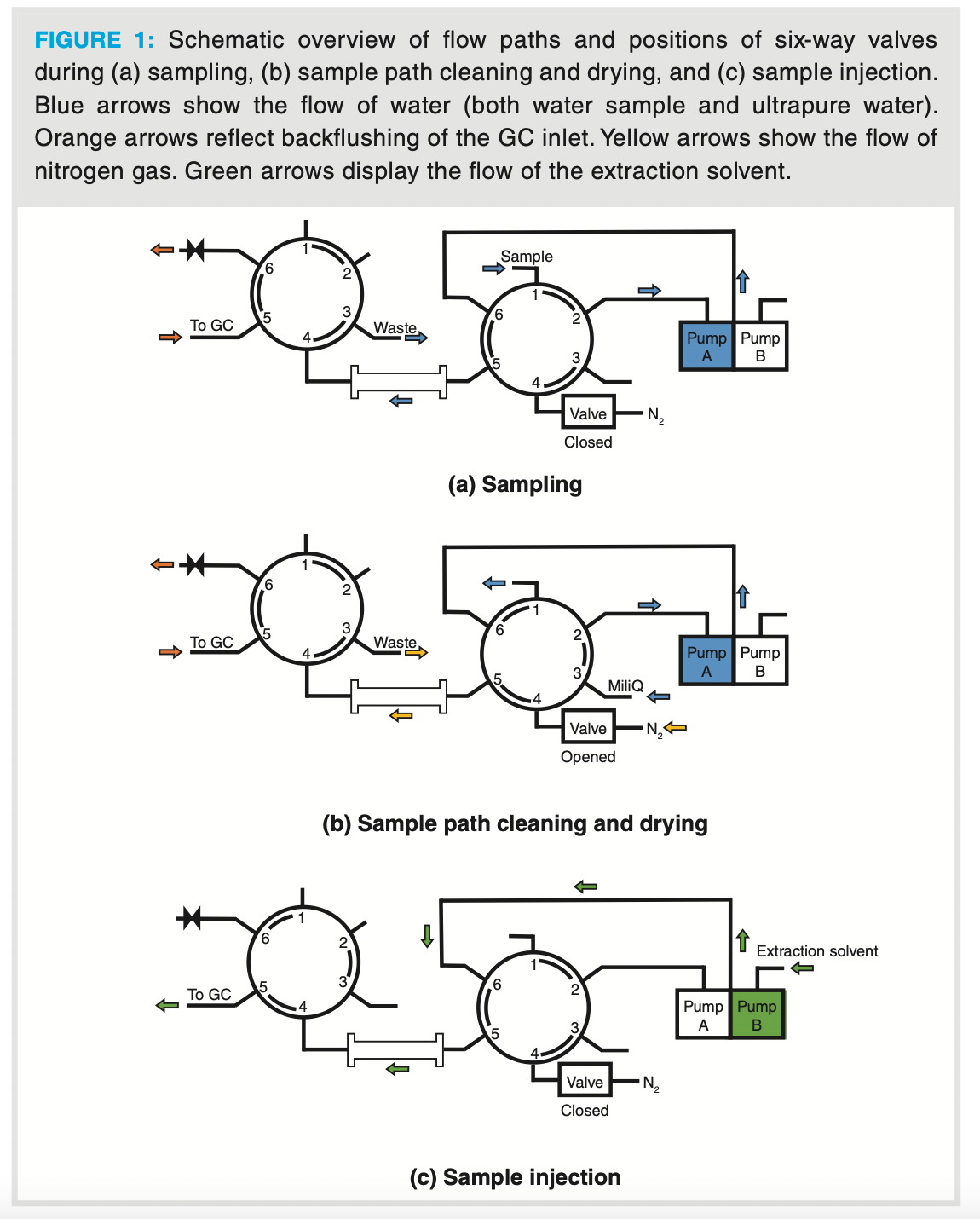
The sampling process was fully automated by a combination of two 6-port rotary valves and a binary pump with wide applicable flow rate range (Figure 1). The aqueous sample was transferred from the sampling bottle over the packed bed via the first 6-port rotary valve. After passing over the packing material, the sample was discarded to waste by means of the second 6-port rotary valve (Figure 1[a]). Several solvents—acetone, hexane, toluene, ethyl acetate—were evaluated to quantitatively recover 2-MIB and geosmin from the extraction bed, and ethyl acetate was selected for the highest recovery in minimal volume. To avoid pressure inconsistencies because of the insolubility of water and ethyl acetate, the packed bed was dry purged with a high flow of nitrogen to remove any residual water prior to administering the ethyl acetate (Figure 1[b]). Afterwards, both 6-port rotary valves changed position to desorb the packed bed (Figure 1[c]). The entire extraction volume (150 µL ethyl acetate) containing the recovered analytes was quantitatively transferred to the GC column.
Results and Discussion
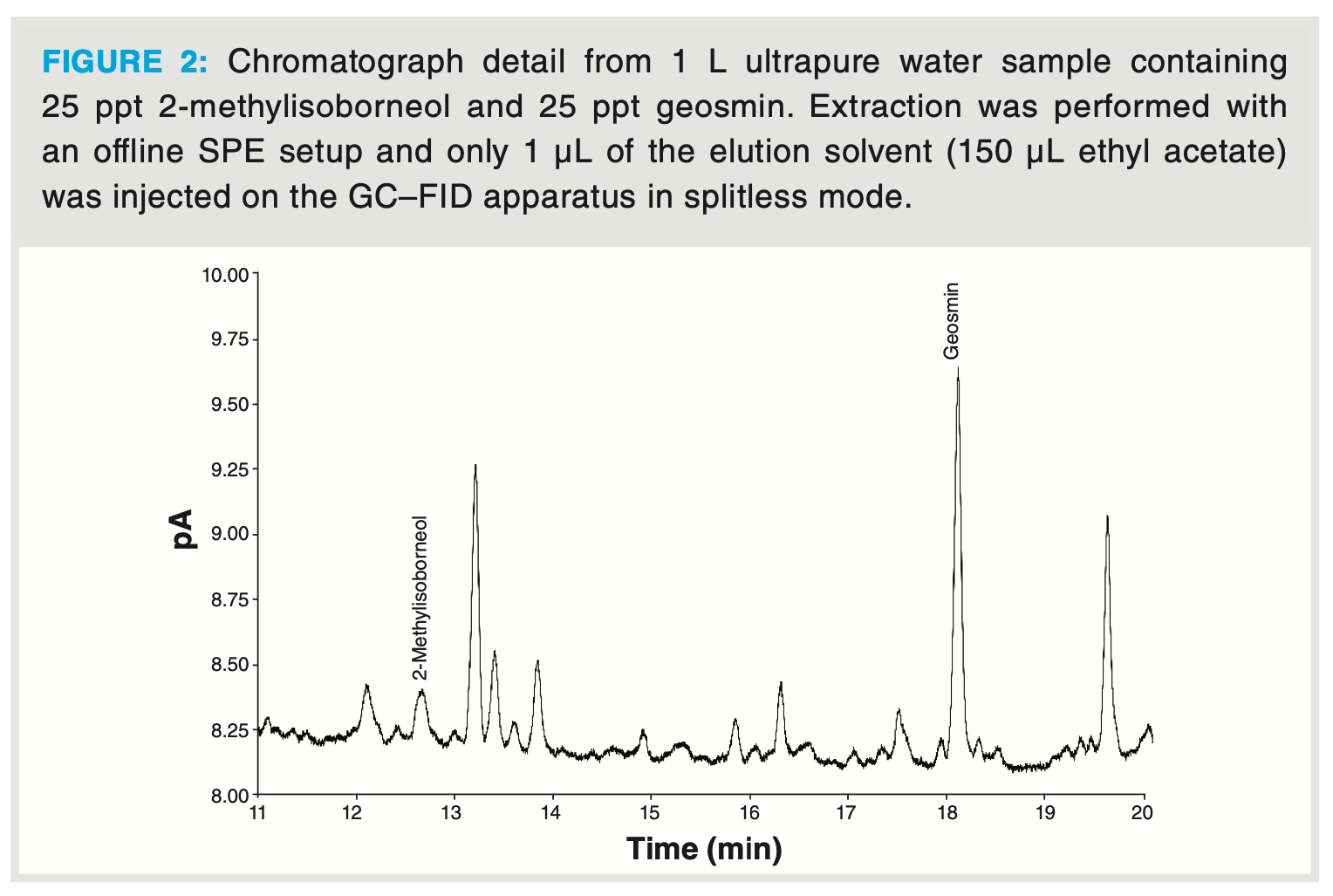
The required limit of detection (LOD) of the online SPE–GC–FID method was maximally 1 ppt. In an initial set of experiments, the method was evaluated in offline mode to evaluate to what extent this LOD could be achieved with reasonable sample volumes. A typical chromatogram resulting from an offline SPE extraction of 2-MIB and geosmin from a 1 L water sample, spiked to a concentration of 25 ppt, is depicted in Figure 2. Ethyl acetate elution volume was 0.15 mL and GC injection volume was 1 µL in splitless mode, which corresponds with 0.16 ng on-column. The corresponding peak for 2-MIB and geosmin were clearly distinguished from the background and achieved a signal-to-noise ratio of 4.4 for 2-MIB and 37.1 for geosmin.

A major benefit of online SPE is that the total volume of ethyl acetate required to recover the trapped components (150 µL) can be introduced onto the GC column, compared with only 1 µL when performing a standard splitless injection, giving rise to the sensitivity gain aimed for. To remove the excess of ethyl acetate prior to analysis, an SVE valve was employed in the setup, meaning that the SVE time is an important parameter that needs careful optimization. If the SVE valve is closed too soon, the amount of ethyl acetate transferred to the analytical column will be far too high, giving rise to column overload and severely disturbed chromatography. On the contrary, if the SVE valve is closed too late, there is a severe risk that not only all the solvent but also all of the analytes of interest will be lost. To determine the optimal SVE time, an offline procedure was applied in which variable volumes of ethyl acetate were introduced using an autosampler rather than using the online setup. The advantages of this approach are apparent. First, disconnecting the SPE module from the GC–FID system allowed the SVE time to be optimized without considering any flow rate restrictions imposed by the system. Very small sample volumes were difficult to generate using the high performance liquid chromatography (HPLC) pump, let alone be transferred to the GC system. Next, improper SVE settings did not severely impact the integrity of the SVE valve, nor did they result in system contamination. SVE time was determined experimentally for injection volumes of 10, 25, 50, and 100 µL. A linear correlation was obtained with a R² of 0.99 between injection volume and SVE time, allowing the SVE time to be extrapolated to 150 µL, that is, the volume of ethyl acetate required for quantitative elution of 2-MIB and geosmin. Final SVE time was 1.3 min. When applied to real samples, the online SPE method exhibited repeatability (n = 6) of 3.13% and 3.93% relative standard deviation (RSD) for 2-MIB and geosmin, respectively, at a concentration of 50 ppt.
By analyzing the entire fraction of extraction solvent, the final concentration of components transferred to the GC–FID system resulted in a LOD below 1 ppt.
Conclusions
An online SPE–GC–FID analyzer is presented for the analysis of ultra‑trace levels of 2-MIB and geosmin in water. The SPE unit allowed concentration factors of 100 to 1000 to be achieved, which allowed common MS technology to be replaced with FID. The online SPE–GC–FID analyzer was compatible with a production environment for online process monitoring.
Water samples were sent over a packed bed by means of a binary HPLC pump. Between sampling and extraction, drying and cleaning steps were carried out to remove any residual water from the bed and the associated tubings. The SVE time was optimized for volumes between 10 and 150 µL, thereby accommodating the injection of the complete extraction solvent.
It was successfully confirmed that the concentrations of odour components 2-MIB and geosmin could be determined using a selective adsorbent bed when present in concentrations below 1 ppt. By direct coupling of the trapping and analysis units, an increase of a factor of 150 was established for the number of recovered components transferred to the GC–FID system.
In future perspectives, other odour components, contaminants, and pollutants will be analyzed with online SPE–GC–FID with selective adsorbents.
Acknowledgements
This work was supported by the Flanders department of agriculture and fisheries (Europees Fonds voor Maritieme Zaken en Visserij [EFMZV] and Financieringsinstrument voor de Vlaamse Visserij- en Aquacultuursector [FIVA]; 19/UP2/06/Aqua) and the KU Leuven Industrial Research Fund (IOF; 3E200929). J.A.M. acknowledges the Flemish government for long‑term structural funding (Methusalem) and the European Research Council (ERC) for an Advanced Research Grant under the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 834134 (WATUSO).
References
- Suurnäkki, S.; Gomez-Saez, G. V.; Rantala-Ylinen, A.; et al. Identification of Geosmin and 2-Methylisoborneol in Cyanobacteria and Molecular Detection Methods for the Producers of these Compounds. Wat. Res.2015, 68, 56–66. DOI: 10.1016/j.watres.2014.09.037
- Watson, S. B.; Monis, P.; Baker, P.; Giglio, S. Biochemistry and Genetics of Taste- and Odor-Producing Cyanobacteria. Harmful Algae 2016, 54, 112–127. DOI: 10.1016/j.hal.2015.11.008
- Darriet, P.; Pons, M.; Lamy, S.; Dubourdieu, D. Identification and Quantification of Geosmin, An Earthy Odorant Contaminating Wines. J. Agric. Food Chem. 2000, 48, 4835–4838. DOI: 10.1021/jf0007683
- Slabizki, P.; Fischer, C.; Legrum, C.; Schmarr, H. -G. Characterization of Atypical Off-Flavor Compounds in Natural Cork Stoppers by Multidimensional Gas Chromatographic Techniques. J. Agric. Food Chem. 2015, 63, 7840–7848. DOI: 10.1021/acs.jafc.5b02793
- Mustapha, S.; Tijani, J. O.; Ndamitso, M. M.; et al. A Critical Review on Geosmin and 2-Methylisoborneol in Water: Sources, Effects, Detection, and Removal Techniques. Environ. Monit. Assess. 2021, 193, 204. DOI: 10.1007/s10661-021-08980-9
- Bristow, R. L.; Young, I. S.; Pemberton, A.; Williams, J.; Maher, S. An Extensive Review of the Extraction Techniques and Detection Methods for the Taste and Odour Compound Geosmin (trans-1, 10-dimethyl-trans-9-decalol) in Water. TrAC – Trend Anal. Chem. 2019, 110, 233–248. DOI: 10.1016/j.trac.2018.10.032
- Ding, Z.; Peng, S.; Xia, W.; et al. Analysis of Five Earthy-Musty Odorants in Environmental Water by HS-SPME/GC-MS. Int. J. Anal. Chem. 2014, 1–11. DOI: 10.1155/2014/697260
- Nakamura, S.; Nakamura, N.; Ito, S. Determination of 2-Methylisoborneol and Geosmin in Water by Gas Chromatography-Mass Spectrometry Using Stir Bar Sorptive Extraction. J. Sep. Sci. 2001, 24, 674–677. DOI: 10.1002/1615-9314 (20010801)
24:8<674::AID-JSSC674>3.0.CO;2-E - Watson, S. B.; Brownlee, B.; Satchwill, T.; Hargesheimer, E. E. Quantitative Analysis of Trace Levels of Geosmin and MIB in Source and Drinking Water Using Headspace SPME. Wat. Res. 2000, 34, 2818–2828. DOI: 10.1016/S0043-1354(00)00027-0
- Mendes, B.; Gonçalves, J.; Câmara, J. S. Effectiveness of High-Throughput Miniaturized Sorbent- and Solid Phase Microextraction Techniques Combined with Gas Chromatography–Mass Spectrometry Analysis for a Rapid Screening of Volatile and Semi-Volatile Composition of Wines—A Comparative Study. Talanta2012, 88, 79–94. DOI: 10.1016/j.talanta.2011.10.010
- Ikai, Y.; Honda, S.; Yamada, N.; et al. Determination of Geosmin and 2-Methylisoborneol in Water Using Solid Phase Extraction and Headspace-GC/MS. J. Mass Spectrom. Soc. Jpn. 2003, 51, 174–178. DOI: 10.5702/massspec.51.174
- Wright, E.; Daurie, H.; Gagnon, G. A. Development and Validation of an SPE-GC-MS/MS Taste and Odour Method for Analysis in Surface Water. Intern. J. Environ. Anal. Chem. 2014, 94, 1302–1316. DOI: 10.1080/03067319.2014.974586
- Fresco-Cala, B.; Jimenez-Soto, J. M.; Cardenas, S.; Valcarcel, M. Single-Walled Carbon Nanohorns Immobilized on a Microporous Hollow Polypropylene Fiber as a Sorbent for the Extraction of Volatile Organic Compounds from Water Samples. Microchim Acta 2014, 181, 1117–1124. DOI: 10.1007/s00604-014-1211-z
- Bulterman, A. -J.; Vreuls, J. J.; Ghijsen, R. T.; Brinkman, U. A. Th. Selective and Sensitive Detection of Organic Contaminants in Water Samples by Online Trace Enrichment–Gas Chromatography–Mass Spectrometry. J. HRC 1993, 16, 397–403. DOI: 10.1002/jhrc.1240160703
- Hankemeier, T.; Louter, A. J. H.; Dallüge, J.; Vreuls, R. J. J.; Brinkman, U. A. Th. Use of a Drying Cartridge in On-Line Solid-Phase Extraction–Gas Chromatography–Mass Spectrometry. J. HRC 1998, 21, 450–456. DOI: 10.1002/(SICI)1521-4168(19980801)21:8<450::AID-JHRC450>3.0.CO;2-J
- Biedermann, M.; Grob, K. On-Line Coupled High Performance Liquid Chromatography–Gas Chromatography for the Analysis of Contamination by Mineral Oil. Part 1: Method of Analysis. J. Chromatogr. A 2012, 1255, 56–75. DOI: 10.1016/j.chroma.2012.05.095
Charlotte Lejaegere is a PhD student at the Centre for Surface Chemistry and Catalysis at KU Leuven University (Leuven, Belgium).
Joeri Vercammen is R&D manager at Interscience Expert Center (Louvain-La-Neuve, Belgium) and visiting professor at Industrial Catalysis and Adsorption Technology at Ghent University (Ghent, Belgium).
Loes Verheyden is a scientist at the Centre for Surface Chemistry and Catalysis at KU Leuven University.
Johan Martens is full professor at the Centre for Surface Chemistry and Catalysis at KU Leuven University.

?")


")



In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




