A New Twist to Ion-Pairing Chromatography: In-Sample Addition of Ion-Pairing Reagent
LCGC North America
Ion-pairing chromatography has traditionally been implemented with the addition of an ion-pairing reagent into the mobile phase. Here, an alternative method is presented, in which the ion-pairing reagent is deposited on column as a sample additive instead of in the mobile phase.
Ion-pairing chromatography has traditionally been implemented with the addition of an ion-pairing reagent into the mobile phase. Here, an alternative method is presented where the ion-pairing reagent is deposited on column as a sample additive and not in the mobile phase. In three separate cases-aminoglycosides, catecholamines (and metabolites), and paraquat–diquat-desirable retention and resolution were achieved on a reversed-phase column with suitable liquid chromatography–mass spectrometry (LC–MS) mobile phases. Three critical factors in achieving desired retention were hydrophobicity, in-sample concentration, and on-column deposition of the ion-pairing reagent. In all cases studied, the ion-pairing reagent exited the column during the cleanup step of the gradient program and could be diverted away to prevent mass spectrometer fouling.
Ion-pairing, paired-ion, and other similarly termed chromatography techniques refer to a venerable technique used for retention and resolution of ionic solutes on a hydrophobic stationary phase (1–7). These terms were assigned based on the perceived behavior between the ionic analytes and a surfactant molecule in the mobile phase within a chromatographic system. The basic operating premise here is the addition of a hydrophobic ion (ion-pairing reagent [IPR]) into the mobile phase to modify the retention characteristics of the stationary phase. This statement presents the most simplistic definition of how ion-pairing chromatography (IPC) works and the IPR's critical function. There has been a significant body of research devoted to ascertaining the retention mechanism of ionic analytes in IPC and, more importantly, the role of the IPR in that mechanism.
Of the three main retention mechanisms that have been put forth (5,8–13), the formation of an ionic–electrostatic layer on the surface of the stationary phase (13–15) is the most widely accepted. Bidlingmeyer observed that the analyte retention is accomplished through a dynamic ion interaction that depends on multiple variables such as temperature, pH, and IPR concentration (13). Cecchi and colleagues have extensively studied this technique and applied rigorous thermodynamic modeling to better explain the retention mechanism and predict a retention equation (16–20). In all of the existing work, mobile phase is used to deliver the IPR onto the stationary phase. This method of delivery may not be the only means to introduce the IPR onto the stationary phase. Liu and colleagues (21) took an unconventional approach by first introducing a large mass of an IPR on a reversed-phase column and proceeded with a subsequent injection of the sample on the column. A gradient method then eluted the analytes and the IPR from the column. More interestingly, Cheng and colleagues reported improved retention of very hydrophilic aminoglycosides antibiotics in protein precipitated human serum samples that contained a high concentration of trichloroacetic acid on a reversed-phase column (22). Although trichloroacetic acid is not commonly regarded as an effective IPR, its impact on improving the hydrophilic analyte retention on reversed-phase columns is noteworthy. To take this matter further, it is interesting to note that if the IPR is introduced only at the head (or a short segment) of the column, many undesirable and problematic attributes of the IPRs in a liquid chromatography (LC) system can be avoided. In general, surfactants and detergents (including IPRs) are among some of the most difficult compounds to remove from a chromatographic system. These molecules are almost universally considered inappropriate for LC–mass spectrometry (MS) applications, mainly because these compounds are not volatile and their sticky nature can cause ion suppression (23,24). Eliminating or significantly reducing the IPR's exposure to an LC–MS system can open the doors to a host of IPC applications in the realm of LC–MS. This notion is investigated in the present work. Three groups of highly hydrophilic basic compounds-aminoglycosides antibiotics, diquat–paraquat herbicides, and catecholamine neurotransmitters (and primary metabolites)-were successfully retained and resolved on reversed-phase LC columns with conventional LC–MS-friendly mobile phases containing no IPR. An acidic group of n-alkyl sulfonates were introduced into the samples to achieve the required on-column retention of the target analytes.
Experimental
Chemicals and Reagents
All compounds in this study were purchased from Sigma-Aldrich. These include uracil (t0 marker); catecholamines and their metabolites (epinephrine, metanephrines, norepinephrine, normetanephrine, dopamine, and 3-methoxytyramine) such as hydrochloride salts or similar; paraquat dichloride and diquate dibromide; and aminoglycoside antibiotics kanamycin sulfate, hygromycin B, gentamicin sulfate (C1 and C2), streptomycin sulfate, dihydrostreptomycin sesquisulfate, neomycin sulfate, and paramomycin sulfate. The ion-pairing reagents sodium n-butyl-, n-pentyl-, n-hexyl-, n-heptyl-, and n-octyl-1-sulfonate salts were also purchased from Sigma-Aldrich. High performance liquid chromatography (HPLC)-grade acetonitrile and methanol were purchased from Thermo Fisher Scientific. Concentrated formic acid was purchased from Sigma-Aldrich. Ultrapure deionized (DI) water was obtained from a Sartorius arium Comfort II system (Sartorius Corporation).
Chromatography
The chromatographic separations of aminoglycosides and paraquate–diquate were carried out on a 100 mm x 2.1 mm, 5-µm dp Phenomenex Kinetex C18 column; catecholamines were resolved on a 50 mm x 2.1 mm, 2.6-µm dp Phenomenex Kinetex F5 column. An Agilent 1260 LC system equipped with a binary pump, autosampler, and column oven was used. Some of the early development was carried out using a Shimadzu Nexera ultrahigh-pressure liquid chromatography (UHPLC) system consisting of two LC30-AD pumps, a SIL30-AC autosampler, and a CTO30-A column oven. A 20-µL static mixer was used to mix the flow from the two LC pumps (Shimadzu America Inc.).
LC Methodology
The aminoglycosides were dissolved in 0.1% formic acid in DI water and further diluted to prepare a 10-µg/mL working standard solution. From this solution, a 300-ng/mL solution was prepared to tune the MS system and conduct chromatographic method development. IPRs were dissolved in DI water to obtain a 0.1 M solution. Aliquots of this solution were used to prepare different concentration of the IPRs in 300-ng/mL aminoglycosides solutions containing an overall concentration of 0.1% formic acid. The mobile-phase solutions used for this method consisted of 0.1% formic acid in water as the weak mobile phase and 100% acetonitrile as the strong solvent (mobile-phase B). The LC gradient started with 5% mobile-phase B and after a 0.5-min delay was increased to 25% mobile-phase B over a 3-min period. After a 1.0-min hold, the proportion of mobile-phase B was increased to 95% in 0.1 min to clean the column. The total chromatographic run time was 8.0 min including the reequilibration period. A constant flow rate of 0.6 mL/min was maintained throughout the LC run.
Similarly, a solution of 10 µg/mL of diquat and paraquat was prepared in DI water and used as a working standard. Subsequently, a solution of 100 ng/mL of these analytes was prepared in 0.1% formic acid in water containing 50 mM n-heptane-1-sulfonate. This mixture was used for the chromatographic analysis. A solution of 0.1% formic acid in methanol (mobile-phase B) was used as the strong solvent for the separation of diquat and paraquat. A solution of 0.1% formic acid in water served as the weak solvent. The LC gradient started with 5% mobile-phase B and was raised to 10% mobile phase B over 5 min. After 0.1 min, the mobile-phase B percentage was raised to 90% over 0.1 min and maintained at that level for 1.5 min. After that time, the gradient was restored to the initial level of 5% mobile-phase B and the column was allowed to reequilibrate. A constant mobile-phase flow rate of 0.6 mL/min was maintained for this analysis.
The catecholamines method was developed on a pentafluorophenyl (Kinetex F5) column with 0.1% formic acid in water as the weak solvent and 0.1% formic acid in methanol (mobile-phase B) as the strong solvent. The LC gradient for this method started out with 5% mobile-phase B and after a 0.5-min delay, the mobile-phase B level was ramped to 50% over a 2.5-min period and held constant for another 0.5 min. In a short step (0.01 min), the mobile-phase B concentration was raised to 95% to clean the column and remove the IPR. Afterward the column was reequilibrated using the initial gradient composition. The mobile-phase flow rate was kept constant at 0.45 mL/min. The entire method run time was 6.5 min.
A solution of 200-ng/mL uracil was prepared in DI water. A second solution containing 200-ng/mL uracil was spiked to contain 50 mM n-heptane-1-sulfonate. Both samples were injected in sequence using the same chromatographic conditions as the probe compounds to determine the t0.
Mass Spectrometry and Detection
The detection of probe compounds and IPRs was carried out on a Sciex Triple Quad 4500 and API 5000 LC–MS/MS system equipped with an electrospray ionization (ESI) probe (Sciex) and operating under multiple reaction monitoring (MRM) scan mode. At least two MRM channels were used for each of the analytes. All probe compounds were positively charged under the experimental conditions and detected in positive polarity with a precursor ion generally consisting of a single charge in the form of [M+H+]. Several of the aminoglycosides produced precursor ions with multiple charges or more-complex ionic forms, such as hydrated and protonated complex ions. In addition, the catecholamines (and metabolites) exhibited dehydration and deamination precursor ions. The MRM transitions along with their respective collision energies are listed in Table I, and compound structures can be found in Figure 1. The IPRs were detected under negative polarity ionization mode on a separate injection using the same mobile phase, flow rate, and gradient program as the probe compounds. Because of the high concentration of the IPRs in the samples, their acquisition methods were detuned to reduce the MS sensitivity while analyzing the IPRs. Generic ion source parameters were used for the ESI in both polarity modes. Data acquisition and reduction were carried out using Sciex Analyst software versions 1.6.1 and 1.6.2.


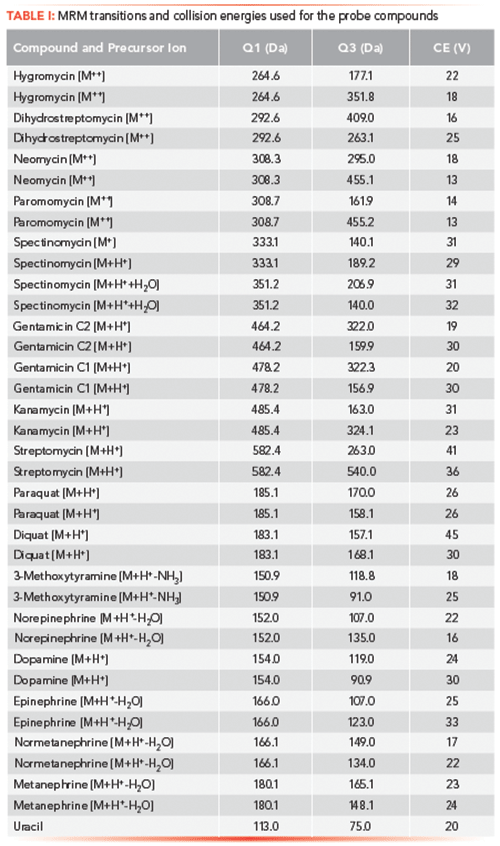

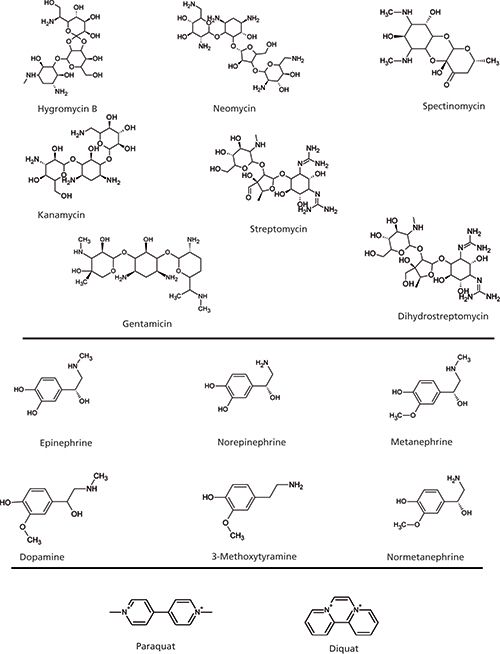
Figure 1: Structure of select probe compounds.
Results and Discussion
Considering that the deposition of the IPR initially occurs at the head of the column during the sample injection, three parameters seemed to be pertinent to establish an effective electrostatic layer inside the column. These parameters are IPR concentration in sample, IPR alkyl chain length (or hydrophobicity), and the amount of IPR deposited on the head of the column. Furthermore, some chromatographic conditions were used specifically to accommodate LC–MS detection. All experiments were carried out under acidic conditions with mobile-phase A consisting of 0.1% formic acid (v/v) in water (pH = 2.7–2.8) and mobile-phase B composed of methanol or acetonitrile. All LC methods were developed to use shallow gradient conditions. During the early investigations, the column temperature played a minor role here and essentially reduced the retention by a small degree for all analytes. With traditional IPC, temperature can play a significant role in affecting the chromatographic selectivity. A possible reason might be the probe compounds' minor structural variations limiting selectivity, especially under the chromatographic conditions used here. This issue will be investigated further, and those findings will be discussed in future work.
Ion-Pairing Reagent Concentration in Sample
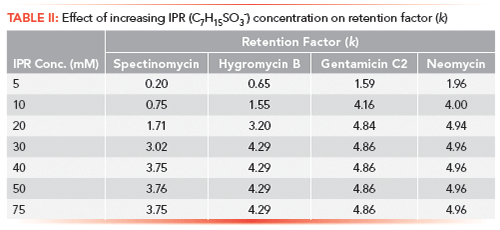
In general, most of the surfactant compounds used here can freely form micelles in an aqueous solution above a certain concentration level, referred to as the critical micelle concentration (CMC). The scope of this study was to use the IPR at concentration levels below their CMC values. The rationales for this decision were, first, to avoid entering the micellar liquid chromatography domain; second, and more importantly, the presence of micelles reduces the availability of the free-form IPR molecules and may interfere with their chromatographic function. The first test performed was to investigate the effect of the IPR concentration in the sample on the retention of the probe compounds on the LC column. For this experiment, sodium n-heptyl-1-sulfonate was chosen as the IPR (CMC = 0.257 M) (25). Individual samples containing 5, 10, 20, 30, 40, 50, and 75 mM of IPR and 300 ng/mL of probe compounds (neomycin, hygromycin B, gentamicin C2, and spectinomycin) were prepared in an acidic (0.1% aqueous formic acid) solution. These samples were injected sequentially on the LC column. However, in-between each sample, a large volume of acetonitrile was injected to remove any trace of IPR from the autosampler injection assembly and the LC column. As the concentration of IPR in the sample increased, so did the probe compounds' retention on the analytical column. Chromatograms of hygromycin B at various concentration of IPR are shown in Figure 2. The increase in the compounds' retention leveled off at an IPR concentration of 40 mM and remained constant at higher IPR concentrations. The calculated retention factors (k) for all probe compounds are listed in Table II. Because of MS detection, it was possible to determine the retention time of n-heptyl-1-sulfonate; it was eluted as the last peak (chromatogram not shown), approximately 1 min after the last probe compound (k = 6.55, retention time [RT] = 3.85 min). Following this data, a concentration of 50 mM n-heptyl-1-sulfonate was used in the samples in all subsequent experiments. The value of t0 was determined with the injection of uracil with and without the IPR in the sample under the same chromatographic conditions, even though the IPR was not expected to affect the uracil retention. In both cases, the t0 values were identical at 0.51 min.

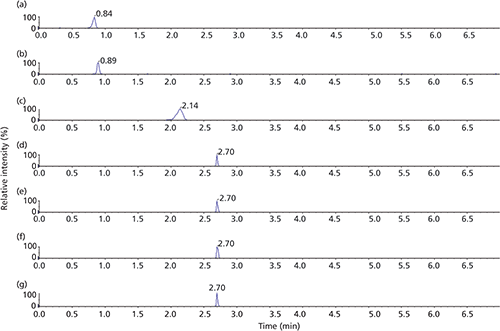
Figure 2: Representative chromatograms of various IPR concentrations on hygromycin B retention. Concentration of n-heptyl-1-sulfonate in the sample: (a) 5 mM, (b) 10 mM, (c) 20 mM, (d) 30 mM, (e) 40 mM, (f) 50 mM, and (g) 75 mM.

IPR Hydrophobicity
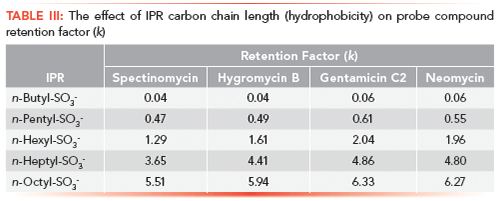
The next criterion to examine was the retention of the probe compounds as a function of the IPR alkyl chain length or IPR hydrophobicity. In traditional IPC, the compound retention is directly proportional to the IPR alkyl chain. Five closely related alkylsulfonate sodium salts were assembled, ranging in length from C4 to C8. The retention factor gradually increased as the number of carbons in the alkyl chain of the IPR increased. Figure 3 represents the effect of alkyl chain length on the retention of hygromycin B. n-Butyl-1-sulfonate appeared to be only slightly effective as we observed breakthrough peaks for most analytes. (Note: The n-butyl sulfonate does not form micelles in aqueous solution and potentially a higher concentration of this reagent could have been used.) The n-octyl-1-sulfonate produced the highest retention of the analytes (see Table III). Interestingly, n-heptyl-1-sulfonate provided the widest elution range for the probe compounds. Based on these data, it was deduced that the alkyl chain length is directly proportional to the analytes' retention.
Figure 2: Representative chromatograms of various IPR concentrations on hygromycin B retention. Concentration of n-heptyl-1-sulfonate in the sample: (a) 5 mM, (b) 10 mM, (c) 20 mM, (d) 30 mM, (e) 40 mM, (f) 50 mM, and (g) 75 mM.

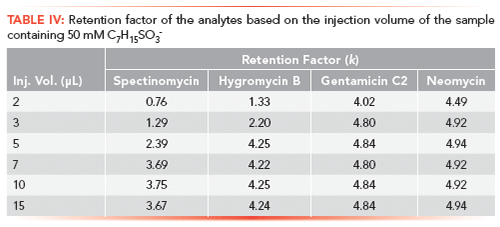

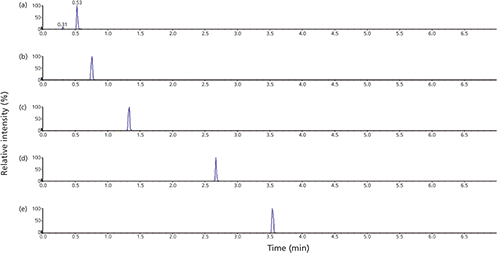
Figure 3: Representative chromatograms showing the effect of IPR alkyl chain length on retention of hygromycin B on column; ion-pairing reagent: (a) n-butyl-1-sulfonate, (b) n-pentyl-1-sulfonate, (c) n-hexyl-1-sulfonate, (d) n-heptyl-1-sulfonate, and (e) n-octyl-1-sulfonate.

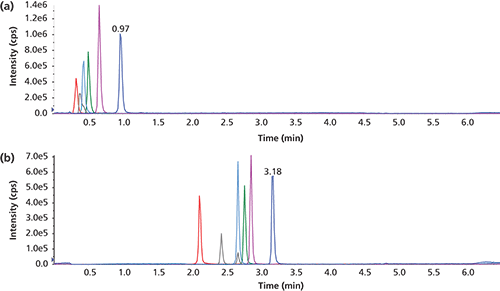
Figure 4: Catecholamines and metabolites separated on a Kinetex F5 column: (a) sample does not contain IPR, (b) sample was prepared in 60 mM n-octyl-1-sulfonate. Peak order: norepinephrine, epinephrine, normetanephrine, dopamine, metanephrines, and 3-methoxytyramine. Note: normetanephrine and epinephrine are isomers and the small peak appearing under normetanephrine is signal collected under the epinephrine MRM transition.

Amount of IPR
Although all LC systems are set up with minimum dead volumes to maximize the chromatographic efficiencies, injected samples may undergo some degree of dilution in the mobile phase. Consequently, it is conceivable that the injected plug should be of sufficient volume (or sufficient IPR mass) to produce an effective area at the head of the column for the IPR to retain the analytes. A set of injections at 2, 3, 5, 7, 10, and 15 µL of probe compounds prepared in 50 mM n-heptyl-1-sulfonate. Similar to the trend for the experiment involving the gradual increase in IPR concentration, the retention for all aminoglycosides increased gradually as injected volume (or deposited mass) increased and then leveled off. The two aminoglycosides with a slightly more hydrophobic nature (gentamicin and neomycin) reached a steady retention at lower injection volumes (Table IV). In all experiments, a minimum injection volume of 5–7 µL seemed to be necessary for a successful retention of all analytes. This volume range represents an on-column deposit of 0.25–0.35 µmol of the IPR on a 2-mm i.d. column.
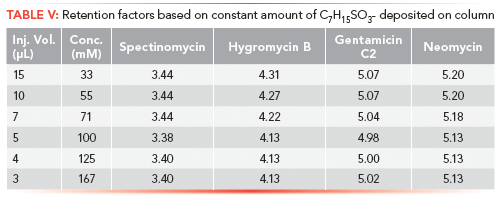
To further clarify the mass and volume relationship, a series of samples was analyzed by depositing the constant mass of IPR on column. However, concentration of the IPR in each sample was progressively larger, and consequently, smaller volumes were injected on the column. These solutions contained n-heptyl-1-sulfonate concentrations of 167, 125, 100, 71, 50, and 33 mM and, respectively, 3, 4, 5, 7, 10, and 15 µL of these solutions were injected on the LC column. The IPR concentration in samples was considered carefully to avoid approaching the CMC of n-heptyl-1-sulfonate (0.257 M). As a result, the 250 mM solution corresponding to a 2-µL injection volume was not attempted. All samples produced fairly similar k values for the four probe compounds (refer to Table V) confirming that the deposited IPR amount is the key factor.

Other Analytes
Catecholamines
The catecholamines and their metabolites consist of epinephrine, norepinephrine, metanephrines, normetanephrine, dopamine, and 3-methoxytyramine. These compounds are among the most difficult groups to chromatographically retain and resolve on reversed-phase columns because of their hydrophilic nature. Unlike the aminoglycosides, these compounds are easily soluble in both aqueous and organic solvents such as methanol. A short-length pentafluorophenyl-based (Kinetex F5) column along with n-octyl-1-sulfonate sample additive was used to retain and resolve these compounds. Figure 4 represents two chromatograms, both obtained with identical column, mobile-phase, and LC gradient program conditions. Figure 4a represents the sample that is prepared in mobile-phase A (0.1% formic acid in water). The sample in Figure 4b contains 50 mM n-octyl-1-sulfonate in 0.1% aqueous formic acid.
Paraquat and Diquat
Paraquat and diquat are herbicides and both contain two quaternary amines in their structure, which makes them excellent candidates for IPC. In a similar fashion as previous examples, mobile-phase A consisted of 0.1% formic acid in water and mobile-phase B was 0.1% formic acid in methanol. The sample contained 50 mM n-heptyl-1-sulfonate. The choice of 0.1% formic acid in methanol as a weaker solvent than acetonitrile produced longer retention and better resolution. A shallow LC gradient program produced retention factors of 2.80 and 3.41 for diquat and paraquat, respectively, and baseline resolution on a C18 column.
Conclusion
Ion-pairing chromatography can be achieved through the addition of an ion-pairing reagent into the sample diluent instead of the mobile phase. Although the mechanism of this new technique has not yet been fully examined, the retention behavior resembles conventional ion-pairing chromatography. For basic compounds, there are several considerations to successfully implement this technique:
- the concentration of the ion-pairing reagent in sample,
- the hydrophobicity of the acidic ion-pairing reagent (alkyl sulfonates), and
- the mass of the ion-pairing reagent deposited on the column.
In three separate classes of compounds-aminoglycosides antibiotics, catecholamine neurotransmitters (and metabolites), and paraquat–diquat herbicides-successful retention and resolution of these compounds were achieved by using IPR as a sample additive under reversed-phase conditions. This technique is specifically beneficial for LC–MS analysis in which the use of nonvolatile mobile-phase components is unacceptable.
References
(1) J.H. Knox and G.R. Laird, J. Chromatogr. 122, 17 (1976).
(2) B. Fransson, K.G. Wahlund, I.M. Johansson, and G. Schill, J. Chromatogr. 125, 327 (1976).
(3) J.C. Kraak, K.M. Jonker, and J.F.K. Huber, J. Chromatogr. 142, 671 (1977).
(4) C.P. Terweij-Groen, S. Heemstra, and J.C. Kraak, J. Chromatogr. 161, 69 (1978).
(5) C. Horvath, W. Melander, I. Molnar, and P. Molnar, Anal. Chem. 49, 2295 (1977).
(6) N.E. Hoffman and J.C. Liao, Anal. Chem. 49, 2231 (1977).
(7) B. Bidlingmeyer, "Bulletin F61," Waters Associates, Inc. (May 1976).
(8) D.P. Witmer, N.O. Nuessele, and W.G. Haney, Jr., Anal. Chem. 47, 1422 (1975).
(9) C. Horvath, W. Melander, I. Molnar, and P. Molnar, J. Chromatogr. 125, 129 (1976).
(10) J.L.M. van de Venne, J.L.H.M. Hendrix, and R.S Deedler, J. Chromatogr. 167, 1 (1976).
(11) P.T. Kissinger, Anal. Chem. 49, 883 (1977)
(12) J.H. Knox, and R.A. Hartwick, J. Chromatogr. 204, 3–21 (1981).
(13) B.A. Bidlingmeyer, S.N. Deming, W.P. Price, Jr., B. Sachok, and M. Petrusek, J. Chromatogr. 186, 419–434 (1979).
(14) F.F. Cantwell, J. Chromatogr. Biomedical Anal. 2(2), 153–164 (1984).
(15) Á. Bartha and J. Ståhlberg, J. Chromatogr. A 668, 255–284 (1994).
(16) T. Cecchi, F. Pucciarelli, and P. Passamonti, Chromatographia 54(9–10), 589–593 (2001).
(17) T. Cecchi, J. Chromatogr. A 958(1–2), 51–58 (2002).
(18) T. Cecchi, F. Pucciarelli, and P. Passamonti, Chromatographia 58(7–8), 411–419 (2003).
(19) T. Cecchi, J. Sep. Sci. 28, 549–554 (2005).
(20) T. Cecchi, Critical Rev. in Anal. Chem. 38, 161–213 (2008).
(21) H. Liu, L. Lam, B. Chi, A.F. Kadjo, and P.K. Dasgupta, Anal. Chem. 88(4), 2059–2064 (2016).
(22) C. Cheng, S. Liu, D. Xiao, and S. Hansel, Chromatographia 72(1–2), 133–139 (2010).
(23) M. Holcapek, K. Volná, P. Jandera, L. Kolárová, K. Lemr, M. Exner, and A. Církva, J. Mass Spectrom. 39(1), 43–50 (2004).
(24) T.M. Annesley, Clin. Chem. 49(7), 1041–1044 (2003).
(25) P.D.T. Huibers, V.S. Lobanov, A.R. Katritzky, D.O. Shah, and M. Karelson, J. Coll. and Interface Sci. 187, 113–120 (1997).
Seyed Sadjadi, J. Preston, and Jeff Layne are with Phenomenex, Inc., in Torrance, California. Direct correspondence to: SeyedS@phenomenex.com



















