My Favorite Shortcuts
Special Issues
Simple ways to estimate chromatographic changes.
Simple ways to estimate chromatographic changes.
There are many equations that allow one to calculate liquid chromatography (LC) parameters-plate number, retention factor, and so forth. If a calculator is required, most of these measurements and calculations are set aside and not made. Yet our work as chromatographers can be simplified, and our methods tend to be better if the method performs within some general limits. Throughout the years, I have learned a number of simple shortcuts, or rules of thumb, that allow me to make quick estimates that are good enough to guide method development or help me solve a problem. This month’s “LC Troubleshooting” will cover a few of these shortcuts to estimating chromatographic parameters.



Retention Factor
The retention factor k is a way of measuring retention in an isocratic separation and can be calculated as:


where tR is the retention time and t0 is the column dead time (“solvent front” time), both in the same units. To get the best chromatographic behavior for isocratic separations, 2 < k < 10 is desired, but sometimes 1 < k < 20 is necessary because of the polarity range of the sample. When k is too small, peaks tend to run together and often are interfered with by the unretained materials that are eluted at t0. When k is too large, run time increases unacceptably and peaks broaden to the point where they are lost in the baseline. Although the calculation of k is simple, it does require locating a calculator, or at least a pencil and paper, so too often the calculation is not made. However, an estimate of k is very simple to make. Examine the components of equation 1. The numerator is simply the corrected retention time; that is, start measuring the retention at t0 instead of the injection point. The denominator is the unit of measure for k. It is easy to visually estimate k by using t0 as a ruler and start measuring retention in units of t0 starting at t0. This is illustrated in Figure 1 where retention is shown in units of both time and t0. In this case, the chromatogram looks nice, but nearly all the peaks are eluted with k < 2. One generally can improve the method performance by increasing k in such cases. This can be done by using a weaker mobile phase (less organic in reversed-phase separations). Because k is not influenced by flow rate, the retention time, now longer, can be compensated for by increasing the flow rate so long as the system pressure limits are not exceeded.

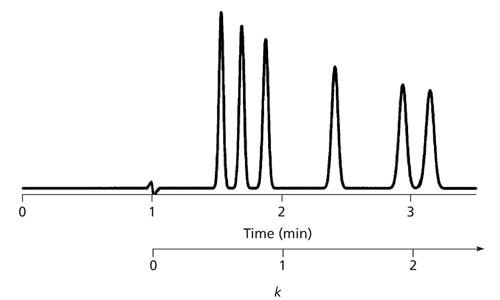
Figure 1: Chromatogram illustrating the use of the t0 ruler as a shortcut for estimating the retention factor. See text for details.
Rule of Three
How much should the mobile phase be changed to increase k the desired amount? Here I like to apply the “Rule of Three,” which states that k (retention) changes about threefold for each 10% change in organic concentration. For example, a 10% reduction in organic would be expected to increase the retention of the first peak of Figure 1 from k ≈ 0.5 to k ≈ 1.5. This would be expected to give a more reliable method because polar materials eluted at t0 would be less likely to interfere with the first peaks in the run. Note that the Rule of Three applies to each 10% change in organic, so a 20% change will change retention 3 x 3 or about 10-fold.
Is t0 Reasonable?
It is a good idea to check t0 in each chromatogram to be sure it is approximately correct. Pump problems, such as check valve malfunction, and leaks will show up as increased values of t0. A shortcut to estimate t0 can be made by first calculating the column volume Vm. For 4.6-mm i.d. columns, this is quite easy:


where L is the column length in centimeters. So a 150 mm (15 cm) x 4.6 mm column will have a column volume of approximately 1.5 mL. This can be converted to t0 by dividing by the flow rate. If the flow rate were 2 mL/min, t0 would be approximately 0.75 min for the present example. This estimate is good to about 10%. If a different column diameter is used, the calculation is a little more complicated:

where dc is the column diameter in centimeters. Note that the column volume is proportional to the square of the column diameter. This makes the change between two of the most common column diameters (4.6 mm and 2.1 mm) very convenient: (4.6/2.1)2 ≈ 5. So rather than try to find your calculator to determine the volume of your 50 mm x 2.1 mm column, just do a mental estimate of its 4.6-mm counterpart (0.1 x 5 ≈ 0.5 mL) and then divide by 5 for a column volume of approximately 100 µL.
What About k for Gradients?
Estimates of k are very useful for isocratic separation, but they don’t apply directly to gradients. This is because the mobile phase composition changes constantly during a gradient run, so k decreases from a large value at the beginning of a run to a small value at the end. An analogous useful estimate of retention in gradient elution can be made with the average k-value, k*:

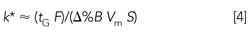
where tG is the gradient time (in minutes), F is the flow rate (in milliliters per minute), Δ%B is the gradient range (in decimal percent; for example, 20% = 0.2), Vm is the column volume (in milliliters), and S is a constant (5 is a good estimate for sample molecular weights < 1000 Da). As with k in isocratic runs, 2 < k* < 10 is a good target for gradient runs. Let’s see how this works for a 20-min full range gradient from 5% to 100% organic using a 150 mm x 4.6 mm column at a flow rate of 1.5 mL/min. Don’t reach for your calculator yet-we can do this one with mental math, too. The numerator of equation 4 is (20 x 1.5) = 30. Δ%B is 0.95, which is close enough to 1 for a quick estimate, so the denominator is (1 x 1.5 x 5) = 7.5. Make the division and you get k* ≈ 4. This tells us that these gradient conditions should give good chromatographic behavior (although no guarantee of the separation we want).
TGIF
Most of us know that TGIF stands for “Thank Goodness It’s Friday.” I also use it to help me make changes in gradient methods. Remember that selectivity α in isocratic separation is defined as the ratio of k-values for adjacent peaks, so if k changes, peak spacing is likely to change also. Changes in mobile phase composition will change retention time, but not t0, so k will change (see equation 1). But changes in flow rate and column volume affect tR and t0 in the same manner, so k stays constant with changes in flow rate or Vm. However, this isn’t true for gradient separations. Equation 3 shows us that k* (and thus selectivity) is affected by flow rate, column volume, gradient time, and gradient range. This means that if the flow rate is doubled, the gradient time must be reduced by half to keep k* the same. It helps to remember that (tG x F) should be kept constant with TGIF (just substitute i for x).
How Much Can I Inject?
Another parameter related to column volume is the guideline for how large an injection can be made. Generally, if the injection is made in mobile phase, you can get away with injecting approximately 15% of the peak volume before noticeable loss in resolution occurs. To estimate the maximum injection volume, first estimate the peak volume. Draw tangents to the sides of the peak and measure the width of the peak at baseline, converting this to volume units. For example, a peak that is 0.5-min wide at 1 mL/min will have a peak volume of 500 µL. This means that you should be able to inject as much as approximately 75 µL of sample dissolved in mobile phase without noticing any problems. If the sample is dissolved in a weaker solvent, a larger volume can be injected, whereas a stronger injection solvent will translate into smaller allowable injection volumes. The best bet is to do an empirical check if you are unsure of the impact of the injection solvent and volume. For example, inject the desired volume and then make injections of half and twice the volume. I like to be able to inject about twice the desired injection volume without peak distortion or retention time shifts.
Column Efficiency
If you read the column test results that are shipped with most columns, you’ll discover that under the manufacturer’s test conditions, a 5-µm dp column will generate 80,000 or more theoretical plates/m. The 3-µm counterpart probably will exceed 100,000 plates/m. This is fine if you are analyzing well behaved hydrocarbon samples, but most of us encounter real-world samples that don’t behave quite as well. Here’s the shortcut that I use to check the column for reasonable performance with my samples:


where N is the number of theoretical plates, L is the column length (in centimeters), and dp is the packing particle diameter (in micrometers). So the 150 mm x 4.6 mm column packed with 5-µm particles should generate approximately 9000 plates (3000 x 15/5). This is about the same as a 10-cm, 3-µm dp column. If the plate number you observe is within about 20% of this value, it is likely that the column is performing properly. If N is significantly less than 80% of this value, it would be good to track down the source of the problem (for example, column failure or extracolumn effects).
Temperature and Retention
An increase in the column temperature has several effects on the LC separation in reversed-phase chromatography. Column back pressure drops because of the decrease in mobile phase viscosity. Retention time is reduced, and generally, peak spacing (selectivity) changes. I use the estimate that retention changes in isocratic separation about 2% for each 1 °C change in temperature. So a 5 °C change in laboratory temperature could reduce retention by 10% if the column were not thermostated. I’ve worked in several laboratories with south-facing windows and poor climate control that could heat up by this much over the course of a sunny summer day. One should always use a column oven with the LC system so that retention times do not drift with changing environmental conditions. Retention changes with column temperature in gradient elution are less pronounced, but changes in selectivity can be significant, so temperature control with gradients also is important.
Keeping Life Simple
Let’s end with a couple of sayings that are useful in guiding method development. The first is the “KISS” principle, which says, “Keep It Simple, Stupid.” This reminds me that the more complicated I make a method, the more problems I am likely to encounter. For example, a binary mobile phase will work more reliably than a ternary one, or a linear gradient will be more reliable than a multisegment gradient. The fewer variables there are to control in a given method, the easier it will be to run.
Second is a saying we use in my laboratory, “Better is the enemy of good enough.” This reminds me to set my goals in advance and when I reach them, don’t spend extra time trying to improve the method. With the use of good method development practices, one often can get a functional separation in a day or so, whereas improving it another 10–20% can take a week or more.
Summary
Like the song in The Sound of Music, “These are a few of my favorite things,” I find that the shortcuts and estimates discussed here are easy and sufficiently accurate for many chromatographic purposes. I’m basically lazy, so if the process requires me to find a ruler and a calculator, chances are that I’ll put it off. On the other hand, if I can make a “guesstimate” of a value, I’ll do it in my head and have the advantage of additional knowledge that can help me troubleshoot a problem or improve a method. I hope you find these useful, too.
How to Cite This Article
J.W. Dolan, LCGC North Am.22(11), 1074–1080 (2004).




















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

