Multidimensional Separation Techniques for Characterization of Biotherapeutics
LCGC North America


Multidimensional separations, in which two or more separation methods are coupled, are a valuable analytical tool for higher peak capacity and improved selectivity for the analysis of complex samples like biotherapeutics.
Multidimensional separations aim to combine two or more separation methods, and these are increasingly being employed to overcome the limitations of one-dimensional separations. The outcome typically includes higher peak capacity and improved selectivity. Such an approach can be invaluable when analyzing complex mixtures. In this article, we discuss some commonly used two-dimensional systems, their pros and cons, and their applications in the biopharmaceutical industry.
The quality of a biotherapeutic is defined by its various biological, immunological, structural, and physicochemical attributes. Some of these are post-translational modifications (PTMs), other product-related heterogeneities, process related impurities, and host cell related impurities. An array of different techniques contributes to the analytical toolbox. These are utilized for characterization of the biotherapeutic product, and especially the myriad of other species that are also present in the final drug product (1–3). One of the most commonly used analytical techniques for routine process evaluations and quality control in the industry is reversed-phase liquid chromatography-mass spectrometry (LC–MS/MS). Due to its robustness, reproducibility, and easy coupling with the MS, LC–MS has become the state-of-the-art technology for routine characterization of biotherapeutics in the industry (3,4). However, LC-based separations suffer from a significant limitation arising from incomplete separation and co-elution of target analytes (5). One reason for lack of baseline resolution is that LC can generate peak capacities up to 1000, and proteolytic digest of large proteins may generate more than 100 peptides. Given the random distribution of peaks, even a system with a peak capacity of 10,000 would be able to resolve only 98% of the mixture of 100 tryptic peptides (6). Thus, in the case of biopharmaceuticals, which are mostly large molecules, use of one-dimensional (1D) chromatography usually does not yield complete, resolved product coverage (6,7).
Multidimensional (MD) separation systems, in which two or more separation methods are coupled, can be used in such cases to increase the peak capacity of the system. This will then allow the complete resolution, it is hoped, of all constituent analytes (8). These systems improve sample resolution manifold by first resolving the components in the first dimension. The eluate is then transferred and further resolved in the second or subsequent dimensions (9). Such a separation was first reported with the use of two-dimensional (2D) paper chromatography in 1944 (8). Since then, different separation techniques have been coupled in a two-dimensional or multidimensional manner, and have been proven useful for the analysis of complex biological samples (9–11).
Based on how the eluate is transferred from one dimension to another, MD separations can be classified as on-line and off-line. The MD system is said to be on-line when the eluate of one dimension is directly transferred to the next dimension, and separation occurs in real time in both the dimensions. On the other hand, in the case of off-line systems, eluates from the first dimension are collected as fractions, and then later subjected to separation in the next dimension (9,12). Both approaches have some advantages and disadvantages (Table I).


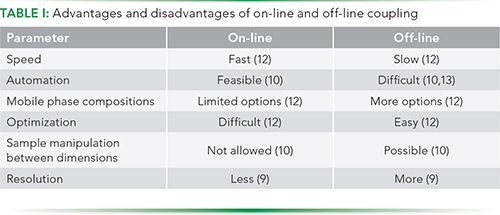
Additionally, MD approaches can also be classified as heart-cutting or comprehensive (1D×2D), depending on the part of the 1D elution content that is transferred in the second dimension. In the heart-cutting MD approach, only a part of the 1D eluate is subjected to separation in the next dimension. However, in a comprehensive approach, the whole eluate of the 1D step is subjected to a 2D separation (9).
Whether on-line or off-line, an MD separation system is useful if it fulfills two fundamental requirements, as suggested by Giddings (8). First, the separation techniques in the MD set up must be orthogonal. This means that the resolving principles of the separation techniques must be based on different chemical or physical properties of the analytes. Thus ideally, for a given analyte, the peak distribution in the two dimensions must be uncorrelated. The second principle states that the resolution of the first dimension must not be lost during separation in subsequent dimensions (8,12).
Multidimensional LC–LC Coupling
One of the most successful MD systems, widely accepted by both academia and industry alike, is that of 2D–LC-based separations (14). These separations involve separation of compounds at two different conditions, using either different modes of LC or using a single-mode operated at two different conditions. Many LC modes, such as ion exchange chromatography (IEC), size exclusion chromatography (SEC) and reversed-phase LC, have been combined. Such couplings have been successfully implemented for the analysis of biopharmaceuticals (11,15). However, not all LC modes can be combined with each other, due to solvent immiscibility and other compatibility issues (16). These 2D-LC systems have been widely accepted by the biopharmaceutical industry. Some of the applications of these techniques in the industry are summarized in Table II.

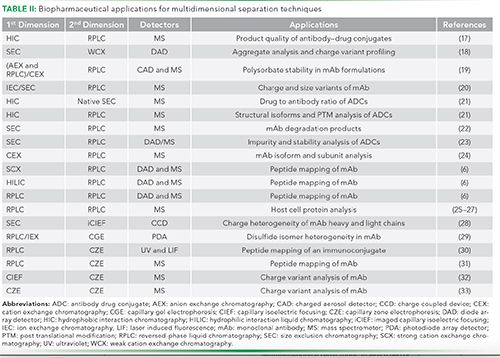
Some of the most commonly used, 2D-LC methods, which have been applied for the characterization of biotherapeutics, are discussed below. In all these methods, reversed-phase LC has been used for the second-dimension separation, likely because of the high-quality reversed-phase LC columns available and their compatibility with MS (34).
Reversed-phase LC x Reversed-phase LC Coupling
This separation system involves resolving the analytes via reversed-phase LC in both dimensions, but at two different pH conditions. The principle is that at high pH (such as pH 10), peptide retention on the reversed-phase LC column is based on solvophobic interactions, while at low pH, both solvophobic and electrostatic interactions determine peptide retention. This difference in peptide retention mechanisms is utilized to constitute the two dimensions of this separation system (35). For instance, researchers have reported an on-line 2D-LC–MS/MS approach for a comprehensive analysis of host cell proteins (HCPs) in monoclonal antibodies (mAbs) (27).
The impact of the use of different cell lines and purification strategies on the composition of HCPs in biopharmaceuticals was analyzed via an on-line, comprehensive 2D-LC–MS/MS analysis. Six PTG1 mAb samples were trypsinized and characterized using MS with high pH reversed-phase LC (pH 10) in the first dimension, and then with low pH reversed-phase LC (pH 2.5) in the second dimension. The 2D-LC system was quite stable, with excellent reproducibility of fractionation in the first dimension, and overall retention times in the second dimension. A reproducibility of 0.1% in retention time was obtained over 48 h of separation, and the reproducibility of the chromatographic setup was independent of the number of fractionations in the first dimension. A total of 33 HCPs were identified over 5 orders of magnitude, with the lowest concentration at 16 ppm. To decrease analysis time and improve peak shape, the second dimension was operated at a high flow rate and high temperature, which then led to a 15% loss in peak capacity as compared to that obtained with standard LC conditions (27). Limited orthogonality is, however, a major issue of this approach (36). Column stability and possible erosion of fused silica tubes at high pH are some of the other factors that must be considered before setting up this separation system (35).
HILIC x Reversed-Phase LC Coupling
One of the best orthogonalities demonstrated is exhibited by the hydrophilic interaction chromatography (HILIC) x reversed-phase LC combination (36). In the first dimension, HILIC separates analytes mainly based on their separation between the mobile phase and water-enriched layer adsorbed onto the stationary phase. In the second dimension, reversed-phase LC separates them on the basis of their hydrophobicity. The use of this setup has been recently reported in a study in which a commercially available 2D-LC system was used for the bottom-up analysis of trastuzumab. A novel dual loop interface was used for transferring the samples between the two dimensions. The use of high elution strength and a high volume of injection solvent, however, led to complete or partial elution of polar peptides in the void volume. Several precautions, such as high flow rate and low temperature of the second dimension, were taken to minimize peptide breakthrough in reversed-phase LC, but it could not be completely eliminated. Unfortunately, the HILIC x reversed-phase LC combination was challenging, and offered limited coverage of the separation space as well. The study compared HILIC x reversed-phase LC with strong cation exchange (SCX) x reversed-phase LC and reversed-phase LC x reversed-phase LC, and it found that the other two techniques are superior for the separation of tryptic peptides of trastuzumab. Although the system offers high orthogonality, concerns persist due to the presence of a high organic content in the mobile phases (35).
SEC x Reversed-Phase LC Coupling
Size-exclusion chromatography (SEC) x reversed-phase LC separates analytes on the basis of size and hydrophobicity in the first and second dimensions, respectively. The system is thus highly orthogonal, and it is useful for peptide separations. Synthetic polymers and biopolymers are some of the major application areas of this system (37). A 2D-LC–MS based, semi-automated platform was developed for characterization of impurities in monoclonal antibodies, with a week-long data collection time. Breakdown products were identified in stress induced mAb samples, using SEC–reversed-phase LC–MS/MS analysis. The low molecular weight breakdown products were enriched by fractionation via SEC in the first dimension, and the fractions were resolved via C8 chromatography in the second dimension. Both on-line and off-line mass spectrometry analyses were performed, using the C8 eluate. A part of the C8 eluate was thus directed to the MS while a major part was collected as fractions. While on-line analysis was used for intact mass analysis, the off-line set up included analysis under both reducing, as well as non-reducing conditions, so as to elucidate impurity structure. Various anticipated products, as well as unpredicted modifications, such as fragment antigen-binding (Fab) fragments and hydrolyzed, free light chain, were identified. The approach was also proposed to be useful for charge variant and aggregation analysis of mAbs.
IEC x Reversed-Phase LC Coupling
In this mode of duality HPLC, ion exchange chromatography (IEC) is used in the first dimension, which separates molecules based on their charge. Analytes are transferred from IEC in the first dimension to reversed-phase LC in the second dimension, as they are eluted with the change in the pH or ionic concentration of the mobile phase. The advantage of using this system is its ease of use, as well as the minimal sample manipulation requirements. Both cation-exchange and anion-exchange chromatography have been coupled to reversed-phase LC to separate protein mixtures and tryptic digests (3,37). For example, rapid analysis of charge variants of mAbs without much sample manipulation was performed, with a generic 2D-LC–MS method. Two independent gradients were used on a single chromatographic system, consisting of two independent column compartments, with built-in switching valves. Reversed-phase trap cartridges were used in the second dimension for desalting first dimension fractions before MS analysis. These cartridges enabled fast conditioning and re-equilibration of columns, with minimal backpressure, during the switching of columns. The poorly resolved and low abundance peaks of IEC in the first dimension, were preconcentrated on the reversed-phase traps using multiple injections. Using an in-process sample of a mAb, fractions of interest were collected from IEC, and then subsequently analyzed via reversed-phase LC, combined with an on-line electrospray ionization (ESI)-time-of-flight (TOF-MS) based detection. Chain specific information about mAbs was also deduced by a disulfide reduction step. This was incorporated as an optional step into the workflow in an on-line manner. Contamination, sample loss, and degradation were minimized with the use of an on-line setup. The whole system was fully automated to allow for high throughput analysis, minimum sample handling, and rapid analysis. The system was shown to be useful for charge, sequence, and chemically unstable mAb variant analysis. Minor glycoforms were also identified at the intact protein level, due to the separation of charged isoforms in the first dimension by IEC (20).
The coupling of SCX in the first dimension and reversed-phase LC in the second is the most widely used 2D-LC system, especially in bottom-up proteomics-based studies. Analytes are separated on the basis of their charge in the first dimension, and hydrophobicity in the second dimension (35). This method, however, suffers from a significant limitation in terms of the peak capacity in the first dimension. The SCX chromatogram is sparsely populated with positively charged tryptic peptides, most of which (+2 and +3 charged peptides) tend to be eluted close to each other. Another major limitation of this method is the limited orthogonality of the system (about 50%). Furthermore, hydrophobic peptides may be eluted with a poor peak shape, or may be incompletely recovered with SCX (36).
The major limitation with these systems, however, is that they show more or less correlated selectivity, and a lower than maximum peak capacity is usually achieved (10,36). Another major limitation of the 2D–LC system is that the separation processes have longer analysis times. Various methods to speed up the LC-based separations via altering column length, temperature, and particle size have been reported. However, each of these methods has its own limitations (37). In 2D-LC, for example, to achieve faster separation, the second dimension LC column is run at high flow rates, which compromises resolution. The second dimension can also be made faster by operating the column at high temperature, which reduces the viscosity of the mobile phase, and allows the user to increase the eluent flow rate, but this may affect the stability of the stationary phase and the analyte (38). Thus, the use of high temperature and other operating parameters, in order to hasten the separation process, often deteriorate the overall, analytical performance of the 2D setup (27).
Multidimensional LC-CE Coupling
Due to the limited orthogonality and other disadvantages associated with LC modes in the second dimension, alternative separation techniques, having different separation principles, have been attempted to improve the separation further. Capillary electrophoresis (CE) is an attractive alternative in this regard, because it separates analytes based on their charge to size ratios, complementary to all modes of LC (10,39). Electrophoresis with chromatography has been described as a powerful 2D-separation method by Giddings (9). The use of CE as the second dimension in 2D systems can thus provide high efficiency and relatively fast separations as compared to LC-based methods. However, special coupling techniques are required for CE based setups, due to the use of small volumes and high electric fields (13). The history of coupling chromatography and electrophoresis dates back to 1948, and since then the coupling has been shown to improve the resolution of various biological and environmental samples (9).
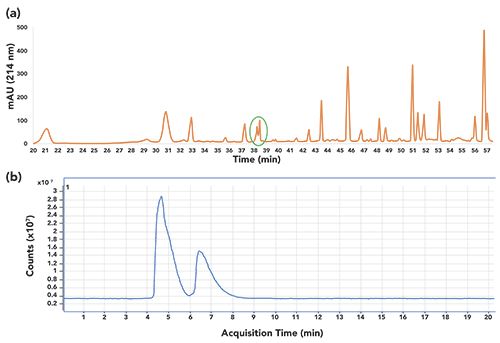
Various methods based on LC–CE, CE–LC, and SEC–CE have been reported in the literature, primarily for proteome and peptide level analysis. An in-depth review of the applications of different modes of CE in MD separations has been recently published (13). The ability of CE to resolve LC peaks has been discussed in the literature (40), and is demonstrated in Figure 1. In this work, the tryptic digest of a protein was resolved using reversed-phase LC in the first dimension, then fractionated, with each fraction subjected to CE–MS analysis in the second dimension. Several overlapping peaks were observed in LC that were efficiently resolved by CE in the second dimension (Figure 1). For instance, the peaks from 38 to 39 min (circled in green in Figure 1a), remained unresolved in reversed-phase LC, as observed in the corresponding UV chromatogram. CE–MS of the corresponding fraction showed baseline resolution of the same peaks (Figure 1b).

Figure 1: Off-line LC-CE-MS of a protein digest. A separation of a protein digest was performed using reversed-phase LC in the first dimension and capillary electrophoresis (CE) in the second dimension. (a) Separation profile of the digest with reversed-phase LC in the first dimension. Solvent A and solvent B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Overlapping peaks at 38–39 min are circled in green. (b) CE–MS profile of the fraction, containing the peaks circled in green in panel a, in the second dimension. CE separation was performed in a 56 cm long polyvinyl alcohol coated capillary at 30 kV. Background electrolyte was 2% acetic acid and detection was performed at 214 nm.
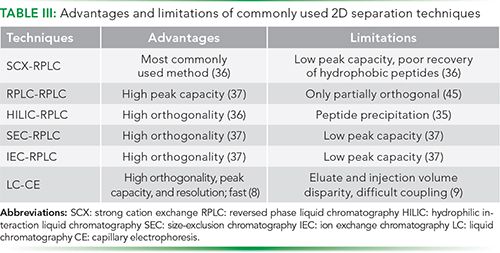
Although these reports clearly demonstrate the utility of LC–CE in biotherapeutics, LC–CE suffers from significant limitations in terms of injection and elution volume disparities, between the LC and CE dimensions. There is also the need to isolate CE from the other parts of the system, due to its high electric field. Due to these reasons, LC–CE has still not gained as much attention as 2D-LC (9). A comparative analysis of the advantages and disadvantages of the commonly used MD methods is given in Table III. Nevertheless, the coupling of LC with CE has been described for analysis of natural compounds, peptide fragments, tryptic peptides of a protein mixture, immunoconjugates of a mAb, and disulfide isomer heterogeneity of IgG2-mAbs (9,10,40–43). For example, a size exclusion chromatography-imaged capillary isoelectric focusing (SEC-iCIEF) method was proposed for the quantitative charge heterogeneity analysis of heavy and light chains of a mAb. Over 10 different mAbs, including both IgG1 and IgG4 subtypes, were characterized using this platform. The platform was found to be less time-consuming, less labor-intensive, more reproducible, and allowed fully quantitative analysis of charge variants compared to the traditional 2D polyacrylamide gel electrophoresis (PAGE) analysis. Results were obtained in a single day, but only with automated SEC and iCIEF platforms. The method was also used for characterization of mAbs under accelerated stability conditions, and it was proposed to be useful for generating simplified electropherograms for conjugated antibodies. It was also useful for obtaining a deeper understanding about the conjugation process. Although this method was well suited for analyzing reduced and denatured mAbs, and offered several advantages over 2D PAGE, it required large amounts of protein sample
for analysis (28).

Four-Dimensional (4D) HPLC-MS
MD setups are not limited to just two- dimensional analysis. Use of more than two dimensions is also emerging for the analysis of biotherapeutics. For instance, recently, an on-line 4D HPLC-MS approach was reported for characterization of mAb variants (44). The mAb sample was resolved and fractionated using IEC in the first dimension. These fractions were then transferred to the 2D reversed-phase column, where they were reduced and washed. The reduced mAb chains were then eluted onto the third column, where they were digested.
The digested peptides were then detected using LC–MS in the fourth dimension. The whole system was fully automated from fractionation to peptide mapping. Prefractionation by IEC allowed reduction and digestion of separated charge variants, resulting in higher signal intensity and sensitivity. The coupling of MS/MS allowed for localizing the modifications at the amino acid level. The system was highly sensitive, and it was able to detect deamidation present, even at 0.5% of the main peak. Five out of six oxidation sites were identified in the study. The system, however, showed limited retention of small and hydrophilic peptides. Although the authors presented IEC in the first dimension, the 4D set-up was designed so that SEC could be coupled in place of the IEC for the characterization of size variants.
Conclusions
Biopharmaceuticals are complex molecules, and they demand complex, analytical procedures with maximum resolving power. To meet this need, one-dimensional analytical methods are not sufficient for achieving complete analysis. Multidimensional systems offer an alternative, promising approach in this regard. 2D-LC is commonly used as a multidimensional separation system for biopharmaceutical analysis, and it has now been accepted by many biopharmaceutical companies. Simplified instrumentation, method development, and data analysis strategies in the future are expected to make it a routine characterization technique for quality control processes in the industry. Various modes are available for 2D-LC systems, but these have limited orthogonality and peak capacity. Recent years have witnessed the growth of CE as a method of choice for MD separation systems. Although these are not yet as widely used as 2D LC-based methods, interest in their deployment is growing rapidly. They represent one of the major developments in the industry which should become more obvious in the future.
References
- A.J. Reason, A. Weiskopf, and A.S. Rathore, BioPharm Intl. 27(7), 1–8 (2014).
- A.S. Rathore, I.S. Krull, and S. Joshi, LCGC North Am.36(6), 376–384 (2018).
- A.S. Rathore, I.S. Krull, and S. Joshi, LCGC North Am.36(11), 814–822 (2018).
- M. Pioch, S.C. Bunz, and C. Neusüß, Electrophoresis33(11), 1517–1530 (2012).
- S.K. Grebe and R.J. Singh, Clin. Biochem. Rev.32, 5–31 (2011).
- G. Vanhoenacker, I. Vandenheede, F. David, P. Sandra, and K. Sandra, Anal. Bioanal. Chem.407(1), 355–366 (2015).
- J.R. Lill, Analytical Characterization of Biotherapeutics (John Wiley & Sons, Hoboken, New Jersey, 1st Ed., 2017) pp. 1–14.
- W. Grochocki, M.J. Markuszewski, and J.P. Quirino, Electrophoresis 36(1), 135–143 (2015).
- L. Ranjbar, J.P. Foley, and M.C. Breadmore, Anal. Chim. Acta950, 7–31 (2016).
- P. Cesla, P. Dinisová, and J. Fischer, Sens. Electroanal.6, 357–368 (2011).
- B.W.J. Pirok, D.R. Stoll, and P.J. Schoenmakers, Anal. Chem.91(1), 240–263 (2019).
- C.R. Evans and J.W. Jorgenson, Anal. Bioanal. Chem.378, 1952–1961 (2004).
- F.J. Kohl, L. Sánchez-Hernández, and C. Neusüß, Electrophoresis36(1), 144–158 (2015).
- X. Wang, S. Buckenmaier, and D. Stoll, J. Appl. Bioanal.3(5), 120–126 (2017).
- H. Wang and S. Hanash, J. Chromatogr. B787, 11–18 (2003).
- P.W. Carr and D.R. Stoll, Two-dimensional Liquid Chromatography (Agilent Technologies, 2015).
- R.E. Birdsall, H. Shion, F.W. Kotch, A. Xu, T.J. Porter, and W. Chen, MAbs 7(6), 1036–1044 (2015).
- S. Schneider, Online 2D-LC Characterization of Monoclonal Antibodies with Size Exclusion and Weak Cation Exchange Chromatography (Agilent Technologies, 2016).
- Y. Li, D. Hewitt, Y. K. Lentz, J. A. Ji, T. Y. Zhang, and K. Zhang, Anal. Chem.86(10), 5150–5157 (2014).
- M. Alvarez, G. Tremintin, J. Wang, M. Eng, Y. Kao, J. Jeong, V.T. Ling, and O. V Borisov, Anal. Biochem. 419(1), 17–25 (2011).
- J.J. Gilroy and C.M. Eakin, J. Chromatogr. B1060, 182–189 (2017).
- M.T. Mazur, R.S. Seipert, D. Mahon, Q. Zhou, and T. Liu, AAPS J.14(3), 530–541 (2012).
- Y. Li, C. Gu, J. Gruenhagen, K. Zhang, P. Yehl, N.P. Chetwyn, and C.D. Medley, J. Chromatogr. A 1393, 81–88 (2015).
- D.R. Stoll, D.C. Harmes, J. Danforth, D. Guillarme, S. Fekete, and A. Beck, Anal. Chem.87(16), 8307–8315 (2015).
- K. Sandra, A. Ortiz, and P. Sandra, LCGC Europe28(10s), 45–48 (2015).
- J.H. Thompson, W.K. Chung, M. Zhu, L. Tie, Y. Lu, N. Aboulaich, R. Strouse, and W.D. Mo, Rapid Commun. Mass Spectrom.28(8), 855–860 (2014).
- C.E. Doneanu, A. Xenopoulos, K. Fadgen, J. Murphy, S.J. Skilton, H. Prentice, M. Stapels, and W. Chen, MAbs4(1), 24–44 (2012).
- R.P. Vanam, M.A. Schneider, and M.S. Marlow, MAbs7(6), 1118–1127 (2015).
- R.K. Roberts, H.J. Holovics, M.R. Thompson, and C.W. Demarest, Electrophoresis31(3), 448–458 (2010).
- J. Liu, H. Zhao, K.J. Volk, S.E. Klohr, E.H. Kerns, and M.S. Lee, J. Chromatogr. A735, 357–366 (1996).
- R. Kumar, R.L. Shah, and A.S. Rathore, J. Chromatogr. A (2020). doi:10.1016/j.chroma.2020.460954
- J. Hühner, K. Jooß, and C. Neusüß, Electrophoresis38(6), 914–921 (2017).
- K. Jooß, J. Hühner, S. Kiessig, B. Moritz, and C. Neusüß, Anal. Bioanal. Chem.409(26), 6057–6067 (2017).
- K. Sandra and P. Sandra, Bioanalysis 7(22), 2843–2847 (2015).
- F. Xie, R.D. Smith, and Y. Shen, J. Chromatogr. A1261, 78–90 (2012).
- M. Gilar, P. Olivova, A.E. Daly, and J.C. Gebler, Anal. Chem.77(19), 6426–6434 (2005).
- D.R. Stoll, X. Li, X. Wang, P.W. Carr, S.E.G. Porter, and S.C. Rutanb, J. Chromatogr. A 1168(1–2), 3–43 (2007).
- M. Kivilompolo, J. Pó, and T. Hyötyläinen, LCGC Europe 24(5), 232–243 (2011).
- D. Chen, X. Shen, and L. Sun, Analyst 142(12), 2118–2127 (2017).
- R. Aebersold and H. D. Morrison, J. Chromatogr. 516, 79–88 (1990).
- S.E. Rudnick, V.J. Hilser, and G.D. Worosila, J. Chromatogr. A 672(1–2), 219–229 (1994).
- H.J. Issaq, K.C. Chan, C. Liu, and Q. Li, Electrophoresis22(6), 1133–1135 (2001).
- E. Tamizi and A. Jouyban, Electrophoresis 36, 831–858 (2015).
- C. Gstöttner, D. Klemm, M. Haberger, A. Bathke, H. Wegele, C. Bell, and R. Kopf, Anal. Chem.90(3), 2119–2125 (2018).
- C. Song, M. Ye, G. Han, X. Jiang, F. Wang, Z. Yu, R. Chen, and H. Zou, Anal. Chem.82(1), 53–6 (2010).
ABOUT THE CO-AUTHOR


Ramesh Kumar is a graduate student at the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.
ABOUT THE COLUMN EDITOR


Ira S. Krull is a Professor Emeritus with the Department of Chemistry and Chemical Biology at Northeastern University in Boston, Massachusetts, and a member of LCGC’s editorial advisory board.
ABOUT THE COLUMN EDITOR

Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.












, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

