Method Development and Validation for Phenol and Nitrophenols in Tap Water by HPLC using a Monolithic Column
LCGC Europe
An isocratic HPLC method for the determination of phenol and nitrophenols (4-nitrophenol, 2-nitrophenol, 4,6-dinitro-o-cresol and 2,4-dinitrophenol) has been developed and validated using 2-chlorophenol as internal standard (IS) and a monolithic column in tap water samples. Prior to HPLC, the method requires solid-phase extraction (SPE) using polymeric Lichrolut EN cartridges. The method development involved the study of methanol and acetonitrile as organic modifiers, pH and flow-rate using a Chromolith RP-18e (150 mm × 4.6 mm I.D.) column. After comparing the performance of the separations obtained with both organic modifiers, the optimum separation of these compounds was achieved using 50 mM acetate buffer (pH 5.0)-acetonitrile (80:20, v/v) as mobile phase, 3 mL min-¹ flow-rate and UV detection at maximum absorbance wavelength. Under these conditions, all analytes were separated (Rs > 2.0) in an analysis time of less than 3.5 min and the most important validation parameters were evaluated. The recoveries obtained in the accuracy test for all phenols studied were in the 90–112% range using a preconcentration factor of 40, and the intraday and interday precisions [expressed as coefficient of variation (CV)] were smaller than 15%. Finally, the proposed method was applied to wastewater samples from several industries.
An isocratic HPLC method for the determination of phenol and nitrophenols (4-nitrophenol, 2-nitrophenol, 4,6-dinitro-o-cresol and 2,4-dinitrophenol) has been developed and validated using 2-chlorophenol as internal standard (IS) and a monolithic column in tap water samples. Prior to HPLC, the method requires solid-phase extraction (SPE) using polymeric Lichrolut EN cartridges. The method development involved the study of methanol and acetonitrile as organic modifiers, pH and flow-rate using a Chromolith RP-18e (150 mm × 4.6 mm I.D.) column. After comparing the performance of the separations obtained with both organic modifiers, the optimum separation of these compounds was achieved using 50 mM acetate buffer (pH 5.0)-acetonitrile (80:20, v/v) as mobile phase, 3 mL min-1 flow-rate and UV detection at maximum absorbance wavelength. Under these conditions, all analytes were separated (Rs > 2.0) in an analysis time of less than 3.5 min and the most important validation parameters were evaluated. The recoveries obtained in the accuracy test for all phenols studied were in the 90–112% range using a preconcentration factor of 40, and the intraday and interday precisions [expressed as coefficient of variation (CV)] were smaller than 15%. Finally, the proposed method was applied to wastewater samples from several industries.
Phenol and nitrophenols are important pollutants in water because of their wide use in many industrial processes as pesticides, insecticides, herbicides and synthetic intermediates.1 Owing to their toxicity both the United States Environmental Protection Agency (EPA) and the European Union (EU) have included some phenols in their lists of priority pollutants. In addition, the 80/778/EC directive states a maximum concentration of 0.5 μg l-1 for total phenols and individual levels under 0.10 mg l-1 for drinking water.
Analytical techniques for phenol determination are mainly high performance liquid chromatography (HPLC) and capillary electrophoresis (CE) using ultraviolet (UV), electrochemical, fluorescence or mass spectrometry (MS) detection.2–3 Gas chromatography (GC), usually after phenols derivatization, has also been used.4 However, it is very difficult, in general, to reach the quantification limits using the above combinations required for the direct determination of phenols in drinking water, and a preconcentration step in the analytical procedure is generally required.
Solid-phase extraction (SPE) and solid-phase microextraction (SPME) are mainly used for phenols preconcentration and for removing interferences from water samples because they provide several advantages over liquid-liquid extraction (LLE). Nevertheless, it is not an easy task because of the different behaviour of the phenols in terms of polarity and acidity. Nowadays, the preconcentration of 500–1000 mL of water with quantitative recoveries for 11 EPA priority phenols would be an important achievement.5
For the SPE of phenols silica-based sorbents, such as C18, C8 and cyclohexil were used. Phenyl and cyano are other silica-based sorbents that have also been used for SPE of phenols in water samples, but none of them give breakthrough volume for phenol higher than 100 mL. Polymeric sorbents based on polystyrene-divinilbenzene (PS-DVB) have higher capacity for polar analytes because of the higher surface area exhibited by polymers and many of the commercially available have areas of 1000 m2 g-1 . In particular, the LiChrolut EN material allows at least 1 L of water with quantitative recoveries for phenol to be concentrated.5
The need for fast, high-resolution separations is sometimes required because of the increase in the number of samples analysed in routine analysis. It has made the columns evolve from a bed packed with porous particles to a straight rod of highly porous silica with a bimodal pore structure (monolithic columns). These columns possess a unique combination of very large internal surface area because of mesopores (13 nm) together with significantly higher total porosity (2 μm macropores) to transport mobile phase and analytes, reducing the diffusion path and providing high permeability (and thus low pressure). This behaviour allows the use of monolithic columns at flow-rates close to 9 mL min-1 without problems and enables faster separations than with a standard column.6–7 In addition, efficiency for monolithic columns does not decrease significantly when the flow-rate is increased because of their flow-through pores, thus diffusion path is reduced, resulting in a reduction in mass transfer effects. However, for traditional particulate columns, using high flow-rates, the efficiency decreases.8
In a previous article, a comparison of the performance of conventional microparticulates and monolithic reversed-phase columns for the HPLC separation of 11 pollutant phenols was reported.9 Taking this study into consideration, an HPLC method for the determination of phenol (P) and some nitro-phenols (NPs) using 2-chlorophenol (2CP) as internal standard (IS) and a monolithic column (Chromolith RP-18e) has been developed. In the method development, the effect of acetonitrile and methanol as organic modifiers, pH and flow-rate have been studied. From the comparison of the performance of the optimal separations obtained using both organic modifiers, acetonitrile was finally selected. This method was validated in tap water samples using SPE LiChrolut EN cartridges. Finally, the proposed method was applied to wastewater samples from automotive, paper mill and petrochemical industries.
Experimental
Chemicals: Phenol (P), 4-nitrophenol (4NP), 2,4-dinitrophenol (24DNP), 2-nitrophenol (2NP) and 2-methyl-4,6-dinitrophenol (46DNOC) and 2-chlorophenol (2CP) were of analytical reagent grade and obtained from Aldrich Chemie (Beerse, Belgium), ). A stock solution of these analytes (1000 μg mL-1 ) was prepared in methanol. A single or a mixture of the phenolic compounds was prepared daily by diluting the stock solution with methanol and used for different studies.
HPLC-grade methanol (MeOH) and acetonitrile (ACN) (Scharlau, Barcelona, Spain) and Milli-Q water (Millipore, Molsheim, France) were used. Millipore 0.45 μm nylon filters (Bedford, Massachusetts, USA) and LiChrolut EN cartridges (200 mg) (Varian, Harbor, USA) were also used. Acetic acid, sodium acetate, sodium dihydrogen phosphate and other reagents were also of analytical reagent grade obtained from Merck (Darmstadt, Germany).
Apparatus: The HPLC system consisted of the following components from Jasco Analítica (Madrid, Spain): a 3-line degasser DG-980-50, a ternary gradient unit LG-980-02S, an HPLC pump PU-980, as well as photodiode array detector (DAD) MD-910. A 6-port Rheodyne valve with a 20 μL sample loop injector (Cotati, California, USA), a Jones Chromatography block heated series 7971 for thermostating columns in the 30–70 °C range (Seagate Technology, Scotts Valley, California, USA) and a Chromolith RP-18e (100 × 4.6 mm I.D.) column from Merck (Darmstadt, Germany) were used.
Mobile phase: Isocratic binary mobile phases were prepared by mixing 50 mM acetate buffer (pH 4.0–5.0) or 50 mM phosphate buffer (pH 3.0) with ACN or MeOH at the required volumes, and the flow-rate was in the 1–4 mL min-1 range and UV absorbance-DAD detection in the 190–360 nm range was also used.
The column temperature was 45 °C and the injection volume was 20 μL. Monitoring the phenols wavelength was adjusted at the maximum absorbance (Table 1).
Sampling and sample pretreatment: Tap water samples were collected from the laboratory. Wastewater samples from petrochemical, automotive and paper mill industries were collected and supplied by environmental persons in charge. Prior to SPE, the water samples were filtered through a 0.45 μm sterile membrane filter (Phenomenex, Torrance, California, USA), were added with HCl to reach pH 2 and stored at 4 °C for further analysis.
Phenol-free tap water samples (PFTW) (with a negative result following the SPE procedure described in the following section) was used as the matrix for phenol spikes before SPE.
Sample preparation: A slightly modified SPE procedure corresponding to reference 10, which uses water samples (100 mL) preconcentrated to a final volume of 10 mL was applied. In this instance, PFTW (200 mL) were spiked with P and NFs in the 5–200 ng mL-1 range and 100 ng mL-1 2CP as internal standard (IS) and processed through LiChrolut EN cartridges (200 mg). The elution was performed by using 2 × 2.5 mL volumes of ACN:MeOH (1:1). Finally, the collected eluate was diluted with Milli-Q water to 5 mL, and 20 μL were injected into the HPLC system. The absolute preconcentration factor was close to 40.
Recoveries, % E, for 11 PPs (including P and NPs) were previously assessed using a developed HPLC method and two different calibration graphs.11 The first one was obtained by direct injection of standard solutions of P and NPs in the 200–8000 ng mL-1 range, and the slope, S1, were calculated in each instance. A second calibration graph was also obtained from PFTW samples spiked with P and NPs in the 5–200 ng mL-1 range after applying the above SPE process, and the corresponding slope, S2, was calculated in each situation. The % E was calculated using Equation 1.



The % E values obtained for P and NPs were higher than 90% and independent of the concentration range used.
Wastewater samples from the above mentioned industries were added with 100 ng mL-1 2CP (IS) and processed in a similar way to PFTW samples.
Results and Discussion
Method development: In a previous article, an optimization of the HPLC separation of a mixture containing 11 phenols using an Hypersil ODS (250 mm × 4.6 mm, i.d. 5 μm) column and methanol (MeOH), acetonitrile (ACN) and tetrahydrofurane (THF) as organic modifiers was performed. After selecting ACN as the most adequate solvent, other relevant chromatographic variables were studied. Under these optimized conditions, several columns including a Chromolith RP-18e (100 mm × 4.6 mm) were tested and the separations obtained were further optimized.9 Temperature for the Chromolith column did not exceed 45 °C owing to its physical properties.

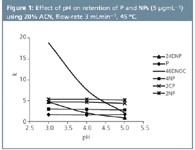
Figure 1
Based on these data, the effect of ACN concentration on P and NPs separation has been studied in the 10–30% range using 50 mM acetate buffer (pH 5.0), a flow-rate of 4 mL min-1 and a Chromolith column (45 °C). As expected, retention factors for these compounds decreased linearly as ACN concentration increased. In this way, all phenols were separated to baseline in the range of 10–20% ACN with analysis times in the 6.0–2.5 min range, respectively.
After selecting 20% ACN, the effect of pH was studied in the 3–5 range. As can be observed in Figure 1, a strong decrease in the retention factor takes place for 46DNOC and 24DNP as the pH increases because of their pKa values (Table 1). In this way, all phenols were separated at pH 4.0 and 5.0 to baseline. From these data pH 5.0 was selected as a compromise between resolution and analysis time.

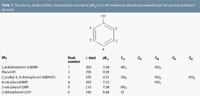
Table 1: Structures, peak number, dissocciation constants (pKa) and UV maximum absorbance wavelength for priority pollutant phenols.
Under the above conditions, the flow-rate was varied in the 1–4 mL min-1 range. A flow-rate of 3 mL min-1 was finally selected, allowing the separation of these compounds to baseline in an analysis time close to 3.5 min with better repeatability than using 4 mL min-1 .
The effect of MeOH concentration on phenols separation was studied (range 20–30%) using the above optimized pH and flow-rate variables. Retention factors for these compounds also decreased linearly as MeOH concentration increases and all phenols were separated to baseline in an analysis time in the 5.5–3.0 min range. An inversion in the elution order for the pair 2CP/2NP was observed as compared with MeOH (range 20–30%) with 20% ACN. From these data 25% MeOH was selected, which allowed the separation of all phenols in an analysis time close to 4 min.
Finally, the performance of the above selected separations using 20% ACN and 25 % MeOH as organic modifiers were compared. For this purpose, several relevant chromatographic parameters such as retention factor, k; selectivity, α; resolution, Rs; asymmetry factors, As; and column efficiency were assessed. Column efficiency was evaluated using the Dorsey-Foley Equation 2

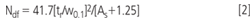
where Ndf is the Dorsey-Foley efficiency, in terms of the asymmetry factor (As) calculated at 10% of the peak height; the retention time (tr) for a given compound and w0.1, the width peak at 10% of the peak height.12 It has been shown that it is a reasonable way to estimate the true efficiency for asymmetric peaks. In addition, plate height (Hdf) values were also calculated from Dorsey-Foley efficiency (Ndf). The values of k, α, Rs, As and Hdf for both separations are summarized in Table 2.
Finally, the separation selected was that obtained using 20 % ACN because this separation shows acceptable As and α values, and the best values for Hdf and Rs with respect to those obtained using MeOH.
Based on this separation, the repeatability as coefficient of variation (CV) was estimated for six standard samples containing 4000 ng mL-1 of each phenol using peak areas (CVa) and tr (CVt). The CVa and CVt obtained were lower than 1%. In summary, the available data obtained from these compounds were adequate enough to develop an analytical method.


Table 2: Chromatographic performance obtained for P and NPs separation using a Chromolith column (45 °C) and ACN (I) or MeOH (II) as organic modifiers.
Analytical Characteristics
Calibration graphs and detection and quantification limits: Standards containing methanolic mixtures of phenol and nitrophenols were prepared in the 200–8000 ng ml-1 range using 5000 ng mL-1 2CP as IS. These mixtures were analysed using 50 mM acetate buffer (pH 5.0)-ACN (80:20, v/v) as mobile phase, a flow-rate of 3 mL min-1 , a Chromolith column (45 °C) and UV-absorbance-DAD at the wavelengths shown in Table 1. The results were analysed by linear regression. Plotting each phenol peak area to IS ratio (PAR) versus the concentration (x) of each one, the calibration equation PAR = Ax + B (μg mL-1 ) was obtained. The A (slope) values were in the 0.0439–0.1296 range, the B (intercept) values were not significantly different from zero and r (regression coefficient) was higher than 0.99 (the errors in the slopes and intercepts do not reveal significant differences). The limits of detection (LODs) and quantification (LOQs) obtained for a signal-to noise (S/N) ratio of 3 and 10, respectively, using calibration graphs were in the 0.96–3.40 ng mL-1 and 3.21–11.33 ng mL-1 ranges, respectively.

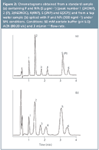
Figure 2
Analysis of Phenol and Nitrophenols in Tap Water Samples using SPE and Method Validation
Calibration graphs, LODs and LOQs: Calibration graphs were obtained by adding to PTFW sample standards of P and NPs at nine concentrations in the 5–200 ng mL-1 range using 100 ng mL-1 2CP under SPE. These mixtures were analysed as described above. In Figure 2 the chromatograms obtained from a standard solution of P and NPs (a) and from a tap water sample spiked with P and NPs (b) under SPE conditions are shown, respectively. Table 3 summarizes the data obtained by linear regression for these compounds (the errors in the slopes and intercepts do not reveal significant differences) and also the LODs and LOQs values. The slope, LOD and LOQ values in Table 3 include the preconcentration factor used in SPE. The calibration equations (Table 3) allow the calculation of phenols concentration levels in tap water samples.


Table 3: Linear regression equations (PAR = Ax + B) using spiked PFTW samples for P and NPs; LODs and LOQs and within- and between-run precision.
Precision (repeatability and reproducibility): Repeatability (Rep) (within-run precision) was examined for P and NPs by analysing 6 different spiked PTFW samples with these compounds under SPE conditions by only one operator within a day. Individual concentrations of 100 ng ml-1 were used, and each sample was run once (n = 6). Reproducibility (Repr) (between run precision) was evaluated for three different days (n = 18). The CV values (CVRep and CVRepr) obtained are shown in Table 3.

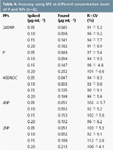
Table 4: Accuracy using SPE at different concentration levels of P and NPs (n=6).
Accuracy: Accuracy was assessed at four different concentration levels of P and NPs by replicate measurements (n = 6). Standards of P and NPs were added to PTFW samples, processed under SPE conditions and analysed using the proposed HPLC method. Table 4 shows the amounts spiked and the recoveries, R (%) and CV, found for P and NPs. These values are higher than 90% and CVs lower than 9.3% in all instances. These results were in agreement with those obtained in a previous SPE assessment of 11 PPs briefly described in sample preparation.11

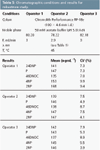
Table 5: Chromatographic conditions and results for robustness study.
Selectivity: Tap water samples were analysed under SPE conditions. A detection and identification process of P and NPs based on tR and DAD was performed. The CV (n = 6) of the retention times for P and NPs was lower than 1% for each one. DAD can provide a contour plot, showing the relationship between absorbance wavelength and time. The UV spectrum of each peak in the chromatogram was stored and subsequently compared with the standards. The spectra were normalized and overlaid. Impurities were studied further by displaying the spectra obtained at different points across the peak. The second derivatives of the spectra across the peak were also obtained to check for peak purity. The possible impurities in each peak were not detected.
Robustness: The robustness test of an analytical method is a measure of its capacity to remain unaffected by small, deliberate variations in method parameters and provides an indication of its reliability during normal usage. Robustness of the proposed method was assessed with respect to alterations in several operational parameters such as the percentage of organic solvent, flow-rate, column temperature and different operators. Tap water samples were analysed by two operators (Nos 2 and 3) (n = 6) using their own standards (150 ng/mL) and under different chromatographic conditions than those used in the present method (Operator No. 1). The working conditions used and the results obtained for the operator are summarized in Table 5. As can be observed, these changes produced acceptable CV values (lower than 10%). These values are within the limits expected for the concentration levels present in these kind of samples, and can be incorporated in the method procedure.13

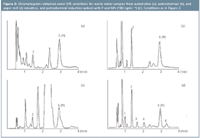
Figure 3
Application to industrial wastewaters: The proposed method was applied to the analysis of P and NPs in different wastewater samples from several industries: automotive (a), petrochemical (b) and paper mill (c). Water samples were analysed as indicated in sampling and sample preparation. Figure 3(a–c) shows the chromatograms obtained for these samples. After checking the retention times of chromatograms and following a detection process similar to that described in the "Selectivity" section. only P was detected in each water sample. Table 6 lists the mean contents (x- ) found (n = 6) along with a statistical data evaluation. These concentration levels are lower than those allowed by the "Comunidad de Madrid" (CM) for these kind of effluents spilled to drainage nets (2000 ng mL-1 ).14


Figure 4
As can be seen in Figures 3(a) and (c) some unknown peaks in these chromatograms show retention times matching with some NPs (Figure 2). However, the behaviour of the petrochemical sample (b) [Figure 3(b)] was not the same. For these reasons, P and NPs (100 ng mL-1 ) were added to the petrochemical sample (b) prior to SPE and analysed as indicated above. Figure 3(d) shows the chromatogram obtained for these compounds, which were detected without interferences. This indicates that the proposed method is potentially adequate to determine these phenols in petrochemical samples. However, for the other kinds of samples it is restricted for some NPs.
Conclusions
The effect of different organic modifiers, pH and flow-rate on the P and NPs separation using a Chromolith column has been studied. Optimal separation was obtained using 50 mM acetate buffer (pH 5.0), 20% ACN and 3 mL min-1 flow-rate. In these conditions all phenols were separated to baseline in an analysis time close to 3.5 min with acceptable As and α values, and the best Hdf and Rs values when compared with those obtained using MeOH as organic modifier. The characteristics of the selected separation have shown to be adequate to develop an analytical method.
The HPLC method was validated for tap water samples under SPE using Lichrolut EN cartridges (the preconcentration factor was close to 40). The validation parameters (linearity, LOD, LOQ, repeatability, reproducibility, accuracy, selectivity and robustness) obtained using SPE have shown to be adequate for the determination of these phenols in tap water samples. The proposed method has been applied to the determination of P and NPs in wastewater samples from automotive, petrochemical and paper mill industries. In all instances, only P was detected. However, the chromatographic behaviour based on retention times and DAD reveals matrix interferences in automotive and paper mill waters with regard to petrochemical samples for some NPs. Finally, petrochemical samples were spiked with P and NPs, which were then detected without interferences. This indicates that the proposed method is potentially adequate to determine these compounds in these kind of samples.
María del Mar Cledera-Castro, PhD, has been an assistant professor in Environmental Science and Materials for Engineering since 1998. She obtained her PhD in 2005. Her main research field is chromatographic method development and its applications to environmental samples. Roberto Izquierdo-Hornillos, PhD, has been a professor in Analytical Chemistry from 1984 and principal scientist from 1990. His main research field is chromatographic method development for drugs and its applications to biological, pharmaceutical, environmental and animal feed matrices. Ana María Santos-Montes is an associate professor in Chemistry for Engineers since 1991. Her main research field is chromatographic method development for drugs and its applications to biological, pharmaceutical and environmental samples.
References
1. L. Zhu, L. Zhu and H.K. Lee, J. Chromatogr. A, 924, 407–414 (2001).
2. A. Peñalver et al., J. Chromatogr. A, 953, 79–87 (2002).
3. J. R.Torres-Lapasió et al., J. Chromatogr. A, 886, 31–46 (2000).
4. M.I. Turnes et al., J. Chromatogr. A, 743, 283–292 (1996).
5. I. Rodríguez, M.P. Llompart and R. Cela, J. Chromatogr. A, 885, 291–304 (2000).
6. N. Tanaka et al., Anal. Chem., 73, 420ª–429A (2001).
7. K. Sinz and K. Cabrera, Environ. Technol., 10(6), 36–37 (2000).
8. D. V. McCalley, J. Chromatogr. A, 965, 51–64 (2002).
9. M. Cledera-Castro, A. Santos-Montes and R. Izquierdo-Hornillos, J. Chromatogr. A, 1087, 57–63 (2005).
10. Application notes 960077 LiChrolut, Chromcircle (V.1.3) Merck KgaA, 64271, Darmstadt, Germany (2004).
11. M. Cledera-Castro, PhD Thesis, Universidad Pontificia Comillas, Madrid, Spain (2005).
12. J. P. Foley and J. G. Dorsey, Anal. Chem., 55, 730–737 (1983).
13. C. Cámara et al., Toma y Tratamiento de Muestras, Editorial Síntesis, Madrid 2002.
14. Official Bulletin Comunidad de Madrid, Law 10/1993, October 26th.









: An Interview With Fabrice Gritti")




, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.


Using GC-MS to Measure Improvement Efforts to TNT-Contaminated Soil
April 29th 2025Researchers developing a plant microbial consortium that can repair in-situ high concentration TNT (1434 mg/kg) contaminated soil, as well as overcome the limitations of previous studies that only focused on simulated pollution, used untargeted metabolone gas chromatography-mass spectrometry (GC-MS) to measure their success.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

Potential Obstacles in Chromatographic Analyses Distinguishing Marijuana from Hemp
April 28th 2025LCGC International's April series for National Cannabis Awareness Month concludes with a discussion with Walter B. Wilson from the National Institute of Standard and Technology’s (NIST’s) Chemical Sciences Division regarding recent research his team conducted investigating chromatographic interferences that can potentially inflate the levels of Δ9-THC in Cannabis sativa plant samples, and possible solutions to avoid this problem.

