Matrix-Assisted Laser Desorption-Ionization Imaging Mass Spectrometry for Direct Tissue Analysis
Special Issues
A summary of the most recent advances in sample preparation, instrumentation, and data-processing techniques for MALDI-IMS
Matrix-assisted laser desorption–ionization (MALDI) imaging mass spectrometry (IMS) is a powerful tool in histopathological characterization and represents a modern analytical technique, enabling two-dimensional detection of molecular components of biological samples. Using this method, it is possible to investigate the spatial distribution of proteins, lipids, carbohydrates, cholesterols, nucleic acids, phospholipids, and small molecules in biological systems by in-situ analysis of cell cultures and tissue sections. Recently, MALDI-IMS has become an essential tool for tissue analyses in life science applications, offering global analysis of tissue samples. An advantage of this imaging technique is the acquisition of local molecular expression profiles up to the microscopic level, while maintaining the topographic integrity of the tissue by avoiding time-consuming extraction, purification, or separation steps, respectively. With MALDI-IMS it is possible to determine the distribution of hundreds of unknown compounds in a single measurement, allowing rapid probing of the tissues' biochemistry. Moreover, MALDI-IMS results include qualitative and semiquantitative information, providing unique chemi-morphological information about the tissue status, which represents an important benefit for future analytical interpretation of pathological changes of a tissue. This article summarizes the most recent advances in sample preparation, instrumentation, and data-processing techniques for MALDI-IMS.
Today, bioanalytical tools based on mass spectrometry (MS) are essential elements in the discovery for new biomarkers and for new drugs that can improve the quality of human life. Rapid assessment of these components' properties is a critical step for enhancing the success rate to find new biomarker or new drug candidates (1). Biomarker discovery is the result of a complex series of comparative proteomics experiments and starts with a diagnosis step on tissue, followed by a differential expression analysis and then marker identification, using orthogonal analytical techniques, among which MS plays a key role (2). In most biomarker discovery and drug discovery studies, high performance liquid chromatography (HPLC)–MS-MS has become one of the standard tools for qualitative and quantitative determination of cell components. However, HPLC–MS-MS is not able to provide answers to certain questions regarding the distribution of certain biomarkers or drugs within individual organs or tissues (3–7). Autoradiography, fluorescence spectroscopy, and immunohistochemistry are still the methods of choice for determining the distribution of certain cell components in organs or tissues. But these techniques have some limitations, because they require the synthesis of radioactive isotopes, fluorescent tags, or specific antibodies, and a direct differentiation between the distribution of an administrated drug and its metabolites is not possible. Over the past few decades a new analytical method, which is generally called matrix-assisted laser desorption–ionization (MALDI) imaging mass spectrometry (IMS) for examination of tissue sections and for visualizing the acquired data, has been introduced. Figure 1 illustrates the MALDI-IMS process, which was described, in parallel to the MALDI profiling procedure, by the laboratory of R.M. Caprioli in 1997 (8).


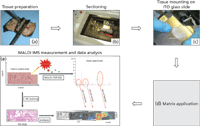
Figure 1: Workflow diagram of sample preparation, MALDI-IMS measurement, and data analysis. Shown are (a) isolation of the medial tissue slice of the prostate specimen; (b) and (c) cutting the prostate sample, after snap-freezing in liquid, and mounting onto ITO glass slides; (d) matrix application with an ImagePrep station; and (e) MALDI-IMS measurement, data analysis, interpretation, and comparison with the histological images.
Today, MALDI-IMS of tissue sections represents an alternative method for the identification of new biomarkers and for the detection of known compounds with no additional labels or affinity tags (9–11). Because of its unmatched sensitivity and high specificity, MALDI-IMS is a powerful tool for the investigation of the spatial distribution of biomarkers in biological samples through the in-situ analysis of thin tissue sections (12,13). Therefore MALDI-IMS, which is a functional microscopic imaging technique, can display the spatial distribution of unlabeled molecules in tissue sections and makes it possible to locate proteins, peptides, lipids, and metabolites by their mass signals in the tissue. MALDI-IMS can contribute to both diagnosis and identification steps. This interplay between those two uses brings a new perspective to the field of biomarker discovery. Technically, this new method uses a soft ionization technique that is capable of transfering intact bio-molecular ions into the gas phase. The result of a MALDI-IMS experiment contains chemical and spatial information (2,13,14). Hence, MALDI-IMS represents a medium-range technique for lateral resolution and sensitivity compared to LC–MS and has a limited dynamic range, mainly as a result of the inability to perform separation techniques on tissue (2,15,16).
MALDI-IMS Tissue Sample Acquisition and Preparation
The chemical representativity and spatial integrity of an IMS experiment depends on sample acquisition, preparation, IMS, and interpretation of the acquired data. In all sample-handling steps throughout the acquisition and preparation process, the spatial and chemical integrity of all compounds must be maintained to avoid delocalization and degradation of the analytes (17). For MALDI-IMS experiments, handling and preparation steps of the sample after procurement are very important to preserve the native condition of the tissue. The main focus should be on the treatment of the tissue specimens peri- and postoperatively, sectioning of the tissue, tissue storage after sectioning, sample transfer to a MALDI target, tissue fixation, and matrix application (18).
Typically, the tissue is snap frozen in liquid nitrogen immediately after tissue collection, conserved at –80 °C, and then cut in a cryostat. Embedding substances such as optimal cutting temperature (OCT) polymer or Tissue-Tek should be avoided to ensure trouble-free measurements, because these embedding materials can smear across the sample during the cutting procedure, resulting in a disturbance of the chemical integrity. For that reason OCT-free cryosectioning of the frozen tissue samples is recommended (19,20). It has also been reported that embedding in gelatin (21) and agarose (19) could be a good alternative to OCT or Tissue-Tek to facilitate handling of small or fragile samples like biopsies. The thickness of the tissue sections is normally between 10 and 20 µm, which represents the optimal thickness for handling and analyzing in the high vacuum environment of the mass spectrometer. Sections thinner than 10 µm can be very fragile and are difficult to manipulate, whereas sections thicker than 20 µm need more time to dry and can adversely affect the performance of the mass analyzer (18). Concerning tissue conservation, MALDI-IMS studies of biological samples do not only involve analysis on fresh frozen-, but also on formalin-fixed tissue. Archived formalin-fixed paraffin-embedded (FFPE) tissue blocks represent a considerable source of information regarding morphological features and molecular content and enable the investigation of the spatial distribution of potential biomarkers. FFPE material has the advantage of preserving molecular features for decades and is compatible with immunochemistry. The main disadvantage of FFPE tissue samples is the formalin induced protein cross-linking, which in general has been assumed to render them unsuitable for proteomic studies. The strategies of analyzing FFPE samples using MALDI-IMS are generally based on in-situ enzymatic digestion of the tissue section after paraffin removal and can be performed on a specific area of the tissue as well as on a very small area. The combination of automated microdigestion of a predefined tissue area with automated microspotting of MALDI matrix according to the same area enables the analysis of the tissue using MALDI-IMS. Nevertheless, the disadvantage of enzymatic digestion treatments in the preparation of FFPE samples is the impairment of an efficient ionization of the proteins and peptides (22,23).
When using frozen material, after cutting the tissue and applying it to the slide, a fixation step before matrix deposition is necessary. One method for fixation is similar to that used in standard histology. First, the tissue is immersed in 70% ethanol, which precipitates the protein content of the tissue, removes salts and impurities, and seems to improve the crystallization process when α-cyano-4-hydroxycinnamic acid (CHCA) is used as matrix. But with the preliminary washing step, a loss of alcohol-soluble compounds can be expected (20). There is also the finding that the removal of lipids by organic solvents such as chloroform, hexane, acetone, toluene, or xylene increases the MS signal of proteins and peptides (24). However, there is no evaluation of chemical modifications, damage, or loss of molecules as a result of these treatments, and no validated technique is available to evaluate the level of molecule delocalization on a biological tissue depending on a particular treatment.
Regardless of whether frozen or FFPE tissue is used, there are two main techniques for tissue analyses using MALDI-IMS: the direct imaging approach and blotting analysis approach, which is less frequently applied (8,25). For the blotting method, targets covered with C18-coated resin beads are used. The first step in this method is the fixation of the blotting membrane on the sample plate. Then, the tissue is blotted onto the membrane, washed with water, dried, and covered with matrix. Preliminary trypsin digestion is also possible. After matrix application, the membrane is analyzed in a mass spectrometer to detect proteins or peptides, but with this technique no direct comparison with the histological features of the measured probe is possible. Given that limitation, the direct imaging approach is more often used, since a direct correlation of MALDI imaging results with the histological features of the probe is possible. On that account, the right choice of the appropriate sample plate is an important aspect for a direct MALDI-IMS experiment. The sample plate of choice is often a transparent conductive slide such as glass slides coated with indium-tin-oxide (ITO) coated, which allows direct correlations between MS images and histological features of the measured tissue section. Glass slides coated by a thin layer of ITO were shown to be compatible with MALDI analysis and with optical microscopy (21,26).
Matrix Selection and Application for MALDI-IMS
The possibility to delocalize molecules on a biological surface efficiently is essential for an IMS experiment and is highly dependent on the matrix depositon procedures. Deposition of a matrix solution can induce delocalization of the compounds in the area covered by the droplet and following crystallization. Therefore, the type of matrix and the deposition process play a preeminent role in a successful MALDI-IMS experiment, and require the choice of the matrix that fits best to the molecular class of interest. The optimum interplay of the type of matrix and the different molecular classes has been determined empirically (27).
For frozen and FFPE tissue analyses, it has been reported that the most used matrices are α-cyano-4-hydroxycinnamic acid (CHCA) and sinapinic acid (SA). SA provides the best signals for higher molecular weight proteins, whereas CHCA is more suitable for lower molecular weight peptides (20). Matrices like 2,5-dihydroxybenzoic acid (2,5-DHB) and 3,4,5-trihydroxyacetophenone (THAP) must be avoided for MALDI-IMS experiments, because they crystallize in the form of long and fine needles, thereby inducing a very significant delocalization of the molecules.
Matrix application protocols for MALDI-IMS include manual methods and automated sample preparation devices. The major disadvantage of manual procedures such as spraying, using an airbrush or thin-layer chromatography (TLC) sprayer, or dipping the tissue sections into matrix-containing solutions is poor reproducibility (20). Higher reproducibility can be achieved using automated sample preparation devices, which include two classes: spotting and spraying devices.
Spotting devices apply small droplets of matrix solution in a grid onto the tissue. In correlation to the actual droplet size, the distance can be enlarged significantly by using a spotting device (28). Spotting devices also can be used to add chemical reagents onto the tissue in a controlled way to perform chemical modifications or digestions on the tissue (29). Disadvantages of robotic spotting procedures are the preparation time, which increases quadratically to the tissue area, and the lateral resolution, which is in the range of 200–500 µm.
Compared to spotting devices, a higher resolution can be achieved by using automated spraying preparation devices such as pneumatic spraying and vibrational spraying devices. Vibrational spraying devices use vibrational vaporization of the matrix with a piezo-electricspray head, which moves a pinhole sheet next to the matrix reservoir to eject small droplets with an average diameter of 20 µm. The matrix aerosol is deposited onto the tissue section under controlled conditions and the entire matrix deposition process is monitored by an optical scattering-light sensor. This sensor monitors the light scattered from matrix crystals to control deposition periods and intervals, matrix layer thickness, wetness, and drying rate in real time. Therefore, this device provides highly reproducible sample preparations for MALDI imaging in a fully automated, push-button process, and represents a significant advantage for routine operation, enabling excellent spectra quality at high image resolution of 25 µm (13). With the pneumatic spraying devices, a constant flow of heated sheath gas is delivered conjointly with the matrix spray, which results in a very rapid evaporation and in consequence minimal analyte delocalization and highly efficient cocrystallization for a superior MS signal. The heated capillary with a pneumatic spray moves in predefined patterns over the tissue sample and sprays with a higher and more reproducible quality compared to a manual pneumatic spray. A disadvantage of this method is the missing control of parameters such as drying rate and wetness of the tissue, which are not monitored during the preparation. Consequently, the settings must be tuned in advance (13).
Histological Staining of MALDI-IMS Tissues
For interpretation of MALDI-IMS results, it is necessary to correlate the allocation of MALDI results with the shape and histological features of the probe. Therefore, there is an urgent need to superimpose the MALDI images onto a macroscopic or microscopic optical image of the sample that was acquired before the MALDI measurement. With a macroscopic optical image it is possible to recognize the outline of the tissue and to define the measurement area, but for the interpretation of the histological features a microscopic image with an appropriate resolution is required. Thus, it is necessary to use stained tissue sections for the analysis of the histological features. However, there are different techniques for the correlation of histology with the MALDI-IMS result: a histological staining of a consecutive section, a histological staining of the measured section before the MALDI-IMS experiment, or a histological staining of the measured section after the MALDI-IMS experiment. With the consecutive-section approach, which allows the use of any staining protocol, one section is used for the MALDI imaging, and another for the histological staining (13). Therefore, hematoxylin-eosin (HE) staining is preferred because it yields a high degree of information about the sample. The HE staining method is popular in histology and is widely used in medical diagnosis. This method involves application of hemalum, a complex formed from aluminium ions and oxidized hematoxylin, which colors the nuclei of cells in blue. The nuclei staining by hemalum does not require the presence of DNA and probably results from binding of the dye–metal complex to arginine-rich basic nucleoproteins such as histones. The hemalum staining is followed by counterstaining with an aqueous or alcoholic solution of eosin Y. Eosin Y colors intracellular or extracellular structures in various shades of red, pink, and orange (30,31). The main difficulty with the consecutive-section approach is the fact that the MALDI image is derived from a different section than the histological image and this only enables a correlation to tissue features with a vague matching. As a result, it remains guesswork if the features seen in the histology are properly matched with the molecular information — at least for exact correlation (13). Histological staining of the measured section before the MALDI-IMS integrates histology and MALDI-IMS and allows an unambiguous correlation of histomorphology and MALDI-IMS. Several commonly used histological dyes such as cresyl violet and methylene blue were tested for compatibility with MS analysis and show a good preservation of spectra quality, whereas HE staining compromises the quality. Disadvantages of this method are the subjection of the sample to additional handling steps before the measurement and the limited choice of stains to those that are compatible with MALDI (26). Therefore, the staining of the sample after the MALDI measurement is the adequate method and allows the use of the best staining for morphological features (HE) and an unambiguous correlation with the MALDI-IMS results (32). Figure 2 shows the histomorphological integrity after MALDI-IMS measurement.


Figure 2: Schematic illustration of the histomorphological integrity after MALDI-IMS measurements. Shown are (a) a frozen sample of a human prostate tissue on a cryostat steel plate; (b) a frozen tissue section (12 µm) mounted onto a conductive glass slide for MALDI-IMS scanned at high resolution; (c) a tissue section after MALDI-IMS measurement (measured region) and MALDI matrix coating on the tissue section; and (d) and (e) the HE stained MALDI-IMS slide. It is observable that the histomorphology after MALDI-IMS is well-preserved as presented on higher magnification in (e).
Visualization and Statistical Analysis of MALDI-IMS Results
Analysis and visualization of MALDI-IMS results are challenging and may become a limitation when a large number of datasets have to be evaluated peak-by-peak. Therefore, many tools have been developed to visualize and analyze these high-dimensional data cubes using various data-processing strategies for visualization. These tools enable fast MS imaging of large surfaces at high-spatial resolution and thus aid in the understanding of various diseases and disorders (13,33,34). The analysis of MALDI-IMS data sets includes region-of-interest analysis, choice of intensity scales for image display (color palette, linear, logarithmic), image overlay, binning of spectra, and image data for improved signal-to-noise, intensity profiles (variation of intensity with time or space), and chemical libraries for analyte identification (17). Available software programs improve the quality of information extracted from the large data sets, reduce the dimensionality to more practical levels, allow different imaging data sets to be aligned and compared, and address data management, including determination of statistical significance and relative abundance between particular protein species. Large numbers of patients, each having unique aspects to their disease, are needed for clinical studies, which points out that individualized medicine is founded on this exciting but enormously complex molecular diversity. Therefore, bioinformatic approaches are necessary to identify individualized molecular patterns that can improve diagnosis and prognosis. It has been reported that data analysis of MALDI-IMS results consists of several steps:
- Selecting proteins that are differentially expressed among histomorphologically predefined groups (for example, healthy and control). The selection of such proteins is based on identifying peaks that significantly differ between the groups using, for instance, statistical significance tests. Such tests include Student's t -test or the non-parametric Wilcoxon rank sum for comparing two groups, and, analysis of variance (ANOVA) and the non-parametric Kruskal-Wallis test for comparing more than two groups (18).
- Reducing the complexity of MALDI-IMS datasets with principal component analysis (PCA), which transforms the original coordinate system defined by peak intensities to a coordinate system that better explains the variance in the dataset. Principal component scores can be used to reconstruct images that contain most of the information, but do not classify the spectra (13).
- Unsupervised classification by hierarchical clustering, which clusters the spectra pair-wise according to similarity until a dendrogram is obtained that contain all spectra. Each branch of the dendrogram can be considered a class of spectra and it is possible to select those dendrogram nodes that reflect certain histological features (35).
In Figure 3, a possible visualization and statistical analysis approach of a MALDI-IMS data set is represented.

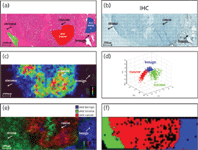
Figure 3: Visualization and statistical analysis of a prostate tissue MALDI-IMS result. Shown are (a) an HE-stained tissue section of human prostate cancer with marked regions of interest (ROIs), in which benign = blue, cancer = red, stroma = green; (b) immunohistochemical validation with marked regions; (c) a MALDI imaging result shown in 2D ion density map representation; and (d) score plots of first, second, and third principal component. Each colored data point of the plot represents one region of interest (ROI). The data sets were chosen from tumor- (red), stroma- (green), and benign tissue region (blue), which are coregistered with the scanned HE slide. This coregistration allows a parallel assessment of the histopathological features and the proteome profiles obtained by MALDI-IMS. (e) The MALDI-IMS result in false color representation. (f) A hierarchical clustering of the human prostate dataset achieved by MALDI-IMS.
MALDI-IMS Used for Proteomics
Several approaches for protein identification of tissue samples, often based on MS tools, coexist in the field of proteomics (36–40). Medical and biomedical proteomics are commonly used to answer three questions:
- How can a specific disease and its stage be diagnosed?
- How can we follow a treatment or evaluate the prognosis?
- What is the mechanism of a disease, the role of protein complexes, and so forth?
The first two questions are the purpose of comparative proteomics in which the nature and concentration of proteins present in "healthy" versus "diseased" samples are compared, especially for biomarker research. The last question is targeted by functional proteomics (41–43). In Figure 4, a classical scheme for biomarker research is illustrated. Batches of samples labeled either "healthy" or "diseased" are separately submitted to protein identification and result in lists of proteins. In the classical approach, resulting protein lists are compared and the proteins that do not match — that is, those that are present, absent, or under- or over-expressed in comparison to the other lists — are potential biomarkers for identification. Identified biomarker candidates then must be confirmed by numerous repetitive experiments by increasing the cohort of samples. After confirmation, the biomarkers must be validated by histological stainings for morphological features and by immunohistochemistry for validation on the protein level by specific antibodies, but such a protocol does not consider the usual high heterogeneity of tissues (2,36). This fact is confirmed for samples diagnosed "diseased," which usually consist of a mixture of healthy and diseased cells without possibility to discriminate. Thus, comparing protein lists from samples labeled "diseased" and "healthy" allows only a selection of biomarker candidates. This limitation can be avoided by laser capture microdissection (LCM). LCM is helpful to select and dissect with a high discrimination between "diseased" and "healthy" samples. But there are limits to the determination of different tissue types, especially at early stages of disease development where only small changes in morphology are present. Other disadvantages of LCM are the necessity of high precision by the operator, the consumption of time, and the small yield of material, which presents a challenge for a full-blown proteomics study (2,44,45).

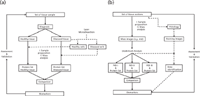
Figure 4: Workflows for biomarker discovery in (a) classical comparative proteomics and (b) MALDI-IMS.
Compared to the classical approach, MALDI-IMS proteomics illustrated in Figure 4, where the analysis is done post-MS by the examination of the tissue distribution of each m/z species, is devoid of prior knowledge and classification. After acquisition of MALDI-IMS results, regions of interest can be defined and analyzed, resulting in classes of compounds. Those masses, which are not homogeneously distributed on defined ROIs, can subsequently be used for tissue classification and identification. In a subsequent step, comparison of generated mass images with images of the experimental sections, stained after MALDI-IMS analysis, allows direct linking the molecular information. Thus, the areas analyzed by MALDI-IMS can be set in correlation to the histopathologically diagnosed tissue type. Hence, the use of MALDI-IMS facilitates the generation of biomarker candidate lists and can be used as a new biomarker discovery tool. The advantage of MALDI-IMS as a biomarker discovery tool is that classification only relies on mass analysis and statistical tools but not on a preliminary diagnosis. The mentioned approaches are capable of detecting many compounds at a time and show great promise in clinical proteomics (2,46,47). However, there are two approaches for protein identification: the top-down approach, which involves ionization and gas-phase fragmentation of the protein of interest inside a mass spectrometer (48), and the bottom-up approach, which uses MS to identify peptides obtained from protease digestion of that protein (49). Resulting mass spectra of the bottom-up approach are searched against theoretical protein–peptide databases for corresponding sequence patterns using conventional algorithms such as Mascot and Sequest (50,51). Incorporating proteomics methodologies into MALDI-IMS protocols, which increases the relevance of this new method to proteomics enhances detection sensitivity and protein identification capabilities. Classical proteomics protocols start with sample collection, followed by sample homogenization, washing, extraction, purification, separation (or separations), enzymatic digestion, further separation steps, preparation for MS analysis, identification of peaks of interest, data interpretation, and validation (52–54). For MALDI-IMS experiments, sample homogenization is not relevant and separation steps like liquid chromatography (LC) or gel electrophoresis must be incorporated for a subsequent identification of peptides and proteins of interest determined by MALDI-IMS. Methods for washing, separation, enzymatic digestion, MS-MS on-tissue and top-down experiments via electron transfer dissociation (ETD) and its related proton transfer reaction (PTR) for identification are possible and have already been shown to improve MALDI-IMS results (2,20,29,55,56). Enzymatic digestion for protein identification coupled with direct tissue analysis involves automatically depositing a spotted array of enzymatic solution onto the tissue at room temperature followed by matrix deposition onto the array for subsequent MALDI-IMS analysis. It has been reported that this approach for the in-situ identification of proteins requires less time than conventional strategies and comparison of the protein image to the image of its subsequent peptides increases identification confidence (29). It also has been demonstrated that direct identification via MS-MS on biological tissues will be a very important tool for MALDI-IMS experiments in the future. The identification potential of MS-MS on biological tissues is used to differentiate between isobaric ions (57–59), to produce specific mass images, usually on targeted molecules like drugs (60), and to identify nontargeted ions on tissues, with the focus on low-molecular-weight ions, especially for specific lipid imaging (57,61). The disadvantage of this technique is that it is not currently sufficient to detect intact proteins. Therefore, a direct analysis strategy of intact proteins has been used. There is increasing interest in this strategy, because of the possibility of a complete protein characterization including termini and modifications. ETD and the corresponding proton transfer (PTR) are considered to be alternative methods for the fragmentation of peptides and proteins. With the ETD-PTR method, it is possible to identify intact peptides and proteins without prior enzymatic digestion. In the first step of this approach, potential biomarker candidates are detected via MALDI-IMS and then are identified via ETD-PTR. Therefore, tissue is extracted and purified and proteins of interest are then identified via ETD-PTR. Such an approach has been tested by Walch, Rauser, and coworkers (55). However, MALDI-IMS can innovate the proteomics world and integrates a comparative proteomics workflow.
Acknowledgments
The authors would like to thank Wolfgang Horninger, Jasmin Bektic, and Viktor Skradsky of the European Prostate Center Innsbruck (Innsbruck, Austria) for providing prostate samples for the biobank. We also gratefully acknowledge Arnd Ingendoh and Konstantin Halikias from Bruker for their aid. We owe thanks also to C. Seifarth from the Department of Urology, Medical University of Innsbruck for sectioning and for assistance with sample preparation. The authors are grateful for the support of the IMGuS project from the Austrian National Foundation for Research, Technology and Development (AWS) and the GENAU project. For support in recording data, we thank M. Becker and S. Schneider, Bruker Daltonik GmbH, Bremen, Germany.
G. Schaefer, H. Klocker, and G. Bartsch are with the Department of Urology at the Medical University of Innsbruck, in Innsbruck, Austria. S. Meding, S. Rauser, and A. Walch are with the Institute of Pathology, Helmholtz Zentrum München, in Neuherberg, Germany. M. Handler, M. Netzer, M. Osl, M. Seger, B. Pfeifer, and C. Baumgartner are with the Institute of Electrical, Electronic, and Bioengineering at the University for Health Sciences, Medical Informatics and Technology, in Hall, Austria. H. Lindner, L. Kremser, and B. Sarg are with the Division of Clinical Biochemistry at the Medical University of Innsbruck, in Innsbruck, Austria. Address correspondence about this article to to C.W. Huck, Head of Spectroscopy Group, at the Institute of Analytical Chemistry and Radiochemistry, Leopold-Franzens University, Innrain 52a, 6020 Innsbruck, Austria, Tel: +43 512 507 5195; Fax: +43 5012 507 2965; or email: Christian.W.Huck@uibk.ac.at.
References
(1) E. Michelini et al., Anal. Bioanal. Chem. 392(3), 355–367 (2008).
(2) L. MacAleese, J. Stauber, and R.M. Heeren, Proteomics 9(4), 819–834 (2009).
(3) M.S. Bryant et al., J. Chromatogr. A 777(1), 61–66 (1997).
(4) B.L. Ackermann, M.J. Berna, and A.T. Murphy, Curr. Top. Med. Chem. 2(1), 53–66 (2002).
(5) G. Hopfgartner, C. Husser, and M. Zell, Ther. Drug Monit. 24(1), 134–143 (2002).
(6) J. Zhang et al., J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 831(1–2), 328–332 (2006).
(7) H.J. Issaq et al., J. Sep. Sci. 32(13), 2183–99 (2009).
(8) R.M. Caprioli, T.B. Farmer, and J. Gile, Anal. Chem . 69(23), 4751–4760 (1997).
(9) Rohner, T.C., D. Staab, and M. Stoeckli, Mech. Ageing Dev. 126(1), 177–185 (2005).
(10) A. Bouslimani et al., Rapid Commun. Mass Spectrom. 24(4), 415–421 (2010).
(11) E.G. Solon et al., AAPS J. 12(1), 11–26 (2010).
(12) D.S. Cornett et al., Nat. Methods 4(10), 828–833 (2007).
(13) A. Walch et al., Histochem Cell Biol. 130(3), 421–434 (2008).
(14) Y. Sugiura, and M. Setou, J. Neuroimmune Pharmacol. 5(1), 31–43 (2010).
(15) P.J. Cassidy and G.K. Radda, J. R. Soc. Interface 2(3), 133–144 (2005).
(16) R.Y. Tsien, Nat. Rev. Mol. Cell Biol. Suppl, SS16–21 (2003).
(17) L.A. McDonnell and R.M. Heeren, Mass Spectrom. Rev. 26(4), 606–643 (2007).
(18) R.L. Caldwell and R.M. Caprioli, Mol. Cell Proteomics 4(4), 394–401 (2005).
(19) R. Kruse and J.V. Sweedler, J. Am. Soc. Mass Spectrom. 14(7), 752–759 (2003).
(20) S.A. Schwartz, M.L. Reyzer, and R.M. Caprioli, J. Mass Spectrom. 38(7), 699-708 (2003).
(21) A.F. Altelaar et al., Anal. Chem. 77(3), 735–741 (2005).
(22) R. Lemaire et al., J. Proteome Res. 6(4), 1295–1305 (2007).
(23) M. Wisztorski et al., Curr. Pharm. Des. 13(32), 3317–3324 (2007).
(24) R. Lemaire et al., Anal. Chem. 78(20), 7145–7153 (2006).
(25) P. Chaurand, M. Stoeckli, and R.M. Caprioli, Anal. Chem. 71(23), 5263–5270 (1999).
(26) P. Chaurand et al., Anal. Chem. 76(4), 1145–1155 (2004).
(27) L.A. McDonnell et al., J. Mass. Spectrom. 40(2), 160–168 (2005).
(28) H.R. Aerni, D.S. Cornett, and R.M. Caprioli, Anal. Chem. 78(3), 827–834 (2006).
(29) M.R. Groseclose et al., J. Mass Spectrom. 42(2), 254–262 (2007).
(30) B.D. Llewellyn, Biotechnic. Histochem. 84(4), 159–177 (2009).
(31) R.D. Lillie, P. Pizzolato, and P.T. Donaldson, Histochem. Cell Biol. 49(1), 23–35 (1976).
(32) K. Schwamborn et al., Int. J. Mol. Med. 20(2), 155–159 (2007).
(33) I. Klinkert et al., Rev. Sci. Instrum. 78(5), 053716 (2007).
(34) T. Villmann et al., Brief Bioinform. 9(2), 129–143 (2008).
(35) G. McCombie et al., Anal. Chem. 77(19), 6118–6124 (2005).
(36) J. Bermúdez-Crespo and J.L. López, Protoemics Clin. Appl. 1(9), 983–1003 (2007).
(37). H. Hondermarck, Pathol. Biol. (Paris) 54(4), 194–198 (2006).
(38) R. Aebersold and D.R. Goodlett, Chem. Rev. 101(2), 269–295 (2001).
(39) B. Domon and R. Aebersold, Science 312(5771), 212–217 (2006).
(40) R. Aebersold and M. Mann, Nature 422(6928), 198–207 (2003).
(41) G.S. Ginsburg and J.J. McCarthy, Trends Biotechnol. 19(12), 491–496 (2001).
(42) M.R. Wilkins, Toxicol. Lett. 127(1–3), 245–249 (2002).
(43) T. Kocher and G. Superti-Furga, Nat. Methods 4(10), 807–815 (2007).
(44) M.R. Emmert-Buck et al., Science 274(5289), 998–1001 (1996).
(45) C. Li et al., Mol. Cell Proteomics 3(4), 399–409 (2004).
(46) R.M. Heeren, Proteomics 5(17), 4316–4326 (2005).
(47) P.G. Righetti et al., Clin. Chim. Acta 357(2), 123–139 (2005).
(48) G.E. Reid and S.A. McLuckey, J. Mass Spectrom. 37(7), 663–675 (2002).
(49) V.H. Wysocki et al., Methods 35(3), 211–222 (2005).
(50) M. Hirosawa et al., Comput. Appl. Biosci. 9(2), 161–167 (1993).
(51) A.J. Link et al., Nat. Biotechnol. 17(7), 676–682 (1999).
(52) B. Canas et al., J. Chromatogr. A 1153(1–2), 235–258 (2007).
(53) A. Bodzon-Kulakowska et al., J. Chromatogr. B Analyt. Techno.l Biomed. Life Sci. 849(1–2), 1–31 (2007).
(54) O. Carrette et al., Nat. Protoc. 1(2), 812–823 (2006).
(55) S. Rauser et al., J. Proteome Res. 9(4), 1854–1863 (2010).
(56) L.H. CazaRes et al., Clin. Cancer Res. 15(17), 5541–5551 (2009).
(57) T.J. Garrett et al., Inter. J. Mass Spectrom. 260(2–3), 166–176 (2007).
(58) P.D. Verhaert et al., Inter. J. Mass Spectrom. 260(2–3), 177–184 (2007).
(59) H. Zhang, S. Cha, and E.S. Yeung, Anal. Chem. 79(17), 6575–6584 (2007).
(60) F.J. Troendle, C.D. Reddick, and R.A. Yost, J. Amer. Soc. Mass Spectrom. 10(12), 1315–1321(1999).
(61) S. Shimma et al., J. Chromatogr. B 855(1), 98–103 (2007).

![Huck_Fig 2-[41825010]-{584590}_t-746305-1408610899253.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Ff6261c79607ac53f41c790ed011e5b2e0d4f0626-200x140.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Huck_Fig 2-[41825010]-{584590}_t-746305-1408610899253.gif")
![Roy figure 5-[41827930]-{585410}-746304-1416911904998.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F5e8a7a8b42be4c1287e42a9e19c61a898804cfe5-700x592.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Roy figure 5-[41827930]-{585410}-746304-1416911904998.gif")
![Schug_Fig 3-[41827280]-{641210}_t-746310-1408610889870.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F6da0479f1e71c57976064cc832407ebe8e770158-200x104.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Schug_Fig 3-[41827280]-{641210}_t-746310-1408610889870.jpg")
![Heinle Figure 1-[41825860]-{582380}_t-746308-1408610893731.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F309898def0ba4426d4cd28df126a523915eb3959-200x103.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Heinle Figure 1-[41825860]-{582380}_t-746308-1408610893731.gif")










New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

