On- and Off-Line Coupling of Separation Techniques to Ambient Ionization Mass Spectrometry
Special Issues
A review highlighting the combination, or potential combination, of various separation methods with ambient ionization mass spectrometry
A review of recent research related to the coupling of ambient ionization mass spectrometry (MS) with various separation techniques is presented. Ambient ionization, referring to a mode of atmospheric pressure ion generation in which sample introduction is segregated from the generation of ionizing radiation under ambient conditions, has generated a great deal of interest in recent years. A plethora of techniques incorporating spray-based and chemical ionization–based source designs have been demonstrated for analysis of analytes from headspace, solution, and solid samples. An overview of some of the more common techniques is given, especially in light of their potential for coupling with different modes of separation, in both on- and off-line formats. This is an emerging area of research that has a strong potential for novel applications, especially given the capability for generating sensitive mass spectral signals from complex samples. In many cases, however, the possibility for coupling efficient separations with current ambient ionization MS exists, but more work remains to comprehensively demonstrate the effectiveness of such hyphenated techniques so that they can be used routinely.
This review highlights the combination, or potential combination, of various separation methods with ambient ionization mass spectrometry (MS) (1). The coupling of separation devices with MS has been recognized as an extremely useful approach for real-world samples analysis, in which complex mixtures are the norm. Typical liquid-phase separation techniques, including high performance liquid chromatography (HPLC), thin-layer chromatography (TLC), and capillary electrophoresis (CE), have been successfully combined with various MS interfaces, such as electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) to create high performance analytical methods (2). Ambient ionization refers to several relatively new ionization techniques that are rapidly advancing toward use in many fields for direct in-situ screening of analytes with minimal or no sample preparation. The popularity of these new ionization techniques is indicated by the explosive growth of the new designs and applications in this area. The ability to separate and independently optimize the sample introduction and the ionizing radiation provides significant advantages compared to more-traditional ionization techniques. Even so, without removing large background interferences, we should be aware that high-resolution and mass-accurate mass detectors are required to unambiguously discriminate the target mass from background noise. This is why additional hyphenation of ambient ionization with separation devices can promote these new techniques for wider applications. Significant advances can be found with these hyphenated techniques, compared with traditional coupling combinations (for example, HPLC and ESI), to a large extent; however, relatively few demonstrations and applications of this promising technology currently exist.
From the standpoint of separation science, factors that can restrict the range of the analysis in current separation–MS combinations still exist in various aspects. For example, nonvolatile buffers such as borate and phosphate that are adopted for better separation in chromatography can cause ion suppression in ESI (3). Also, nonpolar solvents, typically used in normal-phase liquid chromatography, are poorly compatible with ESI due to their low conductivity or low dielectric constants (4). On the contrary, this is another regime where ambient ionization sources could broaden the compatibility between separations and MS. With respect to matrix tolerance and samples present in non-ESI-friendly solvents, some ambient ionization techniques have been shown to provide a higher tolerance to such conditions, compared with traditional atmospheric pressure ionization (5).
Here, we cover the developments and trends of recent research achievements in ambient ionization techniques. Some popular cases will be illustrated with regard to experimental arrangements, mechanisms of ionization, and applications. Following this, some successful combinations between ambient ionization and separation techniques will be narrated. Finally, the potential for this coupling in future work to handle complex sample analysis will be discussed.
Recent Developments in Ambient Ionization
Given the virtue of increased throughput and less stringency in sample form, ambient ionization techniques have become an increasingly useful part of the analytical tool box. Ambient ionization has been widely used in environmental, food, flavor, fragrance, forensic, homeland security, and pharmaceutical areas for trace and real-time analysis (6). A large number of setups have been proposed in the past few years and this effort has been accompanied by an equally long list of new acronyms to remember. Generally speaking, ambient ionization techniques can be classified into two groups based on their associated ionization process. Nominally, these include spray-based (ESI source-modified) and electric discharge-based (APCI source-modified) setups (7–9). Some other variations exist, such as those inspired by atmospheric pressure photoionization (APPI) (10) and a multimode ionization technique that integrates the benefits of both ESI-based and APCI-based ionization techniques (11).
Spray-Based Ambient Ionization
Spray-based ambient ionization techniques often take advantage of conventional ESI to generate charged species, applying it to various situations for gas, liquid, and solid samples analysis, with and without sample pretreatment. Desorption electrospray ionization (DESI) is one of the most prominent and commonly used examples of spray-based ambient ionization techniques. It provides direct solid and liquid sample analysis. The complete design and application was reported by Cooks and coworkers in 2004 (12). The general configuration is shown in Figure 1a. Success with DESI was first demonstrated with small molecules that can be both polar and nonpolar on solid surfaces. The variation of spray solvents (from various aqueous to nonaqueous solvents) plays a critical role for target analyte selection based partly on analyte polarity and solubility (13). Although DESI from solid samples is most common, DESI from liquid samples and flowing liquid samples also has been reported (14).


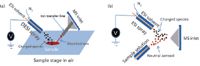
Figure 1: Schematics of popular and established spray-based ambient ionization sources: (a) desorption electrospray ionization (DESI) (adapted from reference 12) and (b) extractive electrospray ionization (EESI) (adapted from reference 29).
A droplet pickup model is hypothesized to be the predominant sampling process in DESI. The sample surface is first wetted by the spray, forming a thin film of liquid on top of the sample. Analytes partition into this thin film and subsequent spray droplets "pick up" the analyte into the ESI droplets, which will continue to desolvate, subdivide, and ultimately lead to release of analyte ions into the gas phase. DESI has been used to analyze a multitude of different analyte types, including nonvolatile pyrolysis products (15), polymer oligomers (16), inks (17), carbohydrates (18), peptides (19), explosives (20–22), human breast cancer tissue (23), porcine and rabbit adrenal glands (24), lipids in mouse brain tissue (25,26), and counterfeit antimalarial drugs (27), among many others (28). Commercial DESI sources are available that are designed to interface to mass spectrometers produced by a variety of manufacturers.
Extractive electrospray ionization (EESI) is another popular spray-based ambient ionization technique that was also introduced by the Cooks group (29). EESI, as shown in Figure 1b, typically is suited for analysis of liquid samples or aerosol samples that can be nebulized. The sample solution is isolated from the ESI source and its high voltage by virtue of its introduction through a second nebulizer. Neutral sample droplets intersect with charged aerosols from the ESI source in front of the MS system inlet (30).
Applications have included the analysis of undiluted urine (29), milk (31), diet effect on biofluids (32), perfume classification (33), and human breath (34), all with minimal or no sample preparation. Zhu (35) and McCullough (36) described the use of EESI for monitoring chemical reactions. Sampling was designed to ensure that the bulk reaction mixture was sampled rather than just the headspace by using a second nebulizer to create an aerosol of the bulk solution using a Venturi pump. The compatibility with liquid samples or aerosols makes EESI a good partner with separation techniques such as liquid chromatography.
In 2008, a modification of the EESI sampling method that uses a neutral desorption (ND) sampling gas beam to gently acquire analytes from sample surfaces, called NDEESI, was proposed by Chen and colleagues (37). This development broadened the EESI application base to trace analysis on biological surfaces and has been demonstrated for use with cheese (38), skin, tissue, muscle, and blood vessels (39).
Chemical Ionization–Based Ambient Ionization
Several significant chemical ionization–based ambient ionization techniques have been reported, including direct analysis in real time (DART) (40), desorption atmosphere pressure chemical ionization (DAPCI) (41), flowing atmospheric pressure afterglow (FAPA) (42), low-temperature probe (LTP) (43), dielectric barrier discharge ionization (DBDI) (44), and microplasmas (45), among others. They basically involve the generation of an electrical discharge across a pair of electrodes in the presence of a flowing gas, such as nitrogen, helium, or vapor in air. Under the high electric field or radiofrequency excitation, a series of ionized molecules, radicals, excited state neutrals, and electrons will be produced that, in turn, can be used to ionize analytes of interest.
DART, schematically depicted in Figure 2, was one of the first ambient ionization sources brought to the market and has been demonstrated for numerous applications, including counterfeit pharmaceutical analysis (46), drug discovery (47), organic reaction monitoring (48), serum metabolite fingerprinting (49), hormone detection (50), and malaria control (51). DART has also been used to directly characterize analytes in conjunction with separations by TLC (52) and HPLC (53).

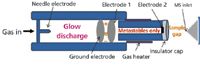
Figure 2: Direct analysis in real time (DART) ion source (adapted from reference 40).
Desorption atmospheric pressure chemical ionization (DAPCI) was first reported in 2005 (41). It was originally investigated as a complementary method to study the ionization process in DESI. DAPCI uses moisture in air or nitrogen to generate the charged species following their passage across a discharge needle. Subsequently, Chen and colleagues successfully applied this technique to the analysis of food, skin, and clothing (54). Additionally, sheath gas is not necessary for good performance in DAPCI, so the analysis of samples in powder form can also be realized (55).
Hyphenation of Ambient Ionization with Various Separation Methods
Ambient ionization (for example, DESI and DART), which provides versatile interfaces between liquid phase separations (such as HPLC) and MS, has been highlighted by recent reports (52,53,56). Significant advantages were demonstrated in terms of the ability to tolerate harsher buffer salts and high flow rates, increase sensitivity and selectivity, and achieve high throughput. These applications are very promising and should help increase interest in coupling ambient ionization with a wider array of separation techniques, in general. In an era where chromatographic separations are being pushed to higher dimensions, the added specificity and versatility of coupling to MS detection through ambient ionization sources will serve to further multiply capabilities. Existing and potential hyphenation of ambient ionization with various separation methods are listed in Table I. Further details and highlights of these achievements and their potential are discussed below. The reader should understand that this may not serve as an exhaustive coverage of this technology.

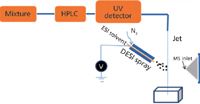
Figure 3: Schematic of a prototype HPLCâDESI-MS apparatus (adapted from reference 61).
Coupling Separations with DESI
The coupling of liquid chromatography with MS through a DESI source was recently demonstrated by Chen and colleagues (Figure 3). Characteristics include a wide range of elution flow rates, online derivatization via reactive DESI (62), and further combination with electrochemistry experiments (61). Three neurotransmitter compounds (3 mg/mL, each), norepinephrine, normetanephrine, and dopamine were separated in isocratic mode at pH 3.0, and fused-silica tubing was used to deliver fractions for DESI analysis. Ionization could be readily generated through the interaction between the charged spray and the HPLC eluent. HPLC flow rates as high as 1.8 mL/min were used in this experiment without a loss of sensitivity.

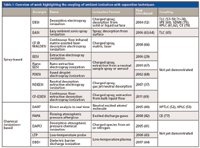
Table I: Overview of work highlighting the coupling of ambient ionization with separation techniques
Besides coupling with HPLC, DESI in combination with TLC is a more natural marriage because of the surface analysis format. Surfaces such as nanoporous silicon, ultrathin-layer chromatography plates, polymethyl methacrylate (PMMA), and polytetrafluoroethylene (PTFE) have been investigated and compared by Cooks and colleagues (70). For all four materials, limits of detection (LODs) were obtained in the femtomole to picomole (picogram to nanogram) range.
Van Berkel and colleagues have taken advantage of this simple and fast method for the direct analysis of gangliosides from TLC plates. Importantly, they demonstrated the potential for automation of this technique by coupling normal-phase TLC with DESI-MS through a computer-controlled scanning device (71). Other applications for the analysis of porcine brain lipids (72) and peptides (73) were also reported. Of course, a significant advantage is that DESI can be used to sample and analyze separation bands without removing or extracting them from the plate. Additionally, the sampling area can be sufficiently small so that the plate can be retained for further analysis by other techniques, if desired.
Similar to a TLC surface, preconcentration and purification techniques such as solid-phase extraction (SPE) and single droplet microextraction (SDME) in which extracted analytes can be enriched on a bead or a frit, respectively, are also amenable to DESI analysis. Direct coupling of SPE with DESI was shown to improve the overall sensitivity up to six orders of magnitude if enough sample was collected (60). Rhodamine 116 was used as the analyte in this study. The lower limit of detection obtained (100 fg/mL–100 pg/mL) was comparable to ESI. For SDME, single aqueous or organic droplets, enriched with trace amounts of methamphetamine (MA) from water, served as a good substrate for DESI analysis (74). The limit of detection (LOD) for MA in the droplet was determined to be 51 ng/mL.
Another classic separation technique is CE. CE is used for separation of ionic species in solution, and recently a unique coupling with DESI was reported by Zare and colleagues (75). Two sets of analyte mixtures, rhodamine dyes and cardiac drugs prepared in high concentrations (10–25 mM) of buffer solutions, were separated by CE and then deposited on a rotating stage (Figure 4a). The researchers were able to show that ion suppression and MS inlet contamination were minimal, despite the high salt concentration. Additionally, the rotating stage format can be used in automated and high-throughput systems.

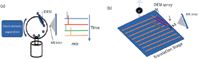
Figure 4: (a) Schematic of capillary electrophoresis coupled with DESI through a rotational spotting/sampling stage (adapted from reference 75). (b) Multichannel device that can be interfaced to DESI for high-throughput analysis (adapted from reference 76).
The capability for increased throughput has always been recognized as an important aspect to consider for a new method to be developed, especially if it is intended for use in industry. To this end, a high throughput device was developed by Ma and colleagues (76) that features a multichannel device with 16 parallel capillaries that can be interfaced with DESI-MS detection (Figure 4b). Of course, the potential for HPLC separations in the capillaries is a significant advantage. The authors indicated that if each sample analysis requires 1.6 min, then an overall throughput of 600 samples/h can be achieved with this technology.
From these examples, it is clear that DESI has enjoyed the most development in terms of interfacing separations with MS detection through an ambient ionization source. Clearly, the ability to sample directly from separation media in various automatable and high-throughput formats, in some cases alleviating potential band broadening effects associated with elution, makes these techniques extremely attractive for use in both routine and high impact research scenarios.
Coupling Separations with DART
Of all of the ambient ionization techniques, DART is second only to DESI in the number of studies in which it has been coupled to various separation formats. Similar to DESI, the coupling of HPLC with DART exhibited significant tolerance to buffer salts in the chromatographic separation (53). Phosphate buffer concentrations as high as 120 mM were used in this combination without evidence of any source contamination or ion suppression. Limits of detection of 20–55 µg/L were achieved for a series of parabens as analytes in the negative ionization mode in this study. The outlet of the HPLC was connected to a stainless steel or fused-silica capillary, and the effluent was directed into the ionizing radiation (Figure 5a). Flow rates as high as 1 mL/min were introduced through the needle into the DART source for MS analysis. Fraction collection is possible by the HPLC-DART combination. After MS analysis, further characterization of the HPLC fractions can be carried out.

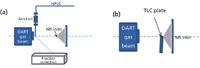
Figure 5: Schematics showing the coupling of (a) HPLC (53) and (b) HPTLC (52) to a DART ion
High performance TLC is also easily coupled with DART-MS (52), as depicted in Figure 5b. Strips of plates containing the separation bands were cut and the edge of each band was exposed to the metastable gas ion beam. Optimal response was observed at a distance of approximately 1 mm from the MS inlet at an angle of about 160° vertical to the gas flow. The coupling was applied in qualitative and quantitative analysis of isopropylthioxanthone. Quantitation was difficult initially, but good linearity (R 2 = 0.9892) and repeatability (RSD < 5.4%, n = 6) was achieved for caffeine detection by introducing an isotopically labeled internal standard. Spatial resolution was shown to be better than 3 mm in this coupling system.
Potential for the Future
EESI (29) and nanoEESI (67), continuous flow infrared matrix-assisted laser desorption electrospray ionization (CF IR MALDESI) MS (66), and continuous flow-extractive desorption electrospray ionization (CF-EDESI) MS (69) have been demonstrated for on-line analysis of liquid samples in recent literature. A natural next step is to couple these with separation techniques, like HPLC and CE. Some work to this end is undoubtedly already being undertaken. Besides, DAPCI, which can ionize an analyte without need for a charged spray and nebulizer gas, holds significant potential for combinations with gas chromatography separations.
Perspective
The capability to independently tune and optimize sample introduction and ionizing radiation in ambient ionization MS holds great promise for further developments in this area. Additionally, since such a high degree of tolerance to solution conditions that hamper traditional API techniques has been demonstrated, the coupling of separations to MS through these ambient ionization sources is immensely attractive. In the earlier years of HPLC, before MS detection was developed into a routine approach, many separations were developed that featured high concentrations of nonvolatile salts. It appears that these high quality separations could now be revisited. As separations advance to higher complexity and dimensionality, the fewer the limitations with detection, the better. With robustness, versatility, and increased throughput capabilities, it can be expected that the number of studies featuring the combined use of high efficiency separations in virtually all phases and formats with the vast array of ambient ionization techniques continually being developed, will amass at a significant rate.
Li Li and Kevin A. Schug are with the Department of Chemistry & Biochemistry at the University of Texas at Arlington.
References
(1) G.A. Harris, A.S. Galhena, and F.M. Fernandez, Anal. Chem. 83, 4508–4538 (2011).
(2) K.B. Tomer, Chem. Rev. 101, 297–328 (2001).
(3) J.P. Antignac, K.D. Wasch, F. Monteau, H.D. Brabander, F. Andre, and B.L. Bizec, Anal. Chim. Acta. 529, 129–136 (2005).
(4) N.B. Cech and C.G. Enke, Mass Spectrum. Rev. 20, 362–387 (2001).
(5) H. Chen, G. Gamez, and R. Zenobi, J. Am. Soc. Mass Spectrom. 20 , 1947–1963 (2009).
(6) F.M. Green, T.L. Salter, P. Stokes, I.S. Gilmore, and G. O'Connor, Surf. Interface Anal. 42, 347–357 (2010).
(7) M. Huang, C. Yuan, S. Cheng, Y.T. Cho, and J. Shiea, Annu. Rev. Anal. Chem. 3, 43–65 (2010).
(8) G.A. Harris, A.S. Galhena, and F.M. Fernandez, Anal. Chem. 83, 4508–4538 (2011).
(9) H. Chen, B. Hu, and X. Zhang, Chin. J. Anal. Chem. 38, 1069–1088 (2010).
(10) M. Haapala, J. Pól V. Saarela, V. Arvola, T. Kotiaho, R.A. Ketola, S. Franssila, T.J. Kauppila, and R. Kostiainen, Anal. Chem. 79, 7867–7872 (2007).
(11) L. Nyadong, A.S. Galhena, and F.M. Fernandez, Anal. Chem. 81, 7788–7794 (2009).
(12) Z. Takáts, J.M. Wiseman, B. Gologan, and R.G. Cooks, Sci. 306, 471–473 (2004).
(13) A.B. Tawiah, C. Bland, D.I. Campbell, and R.G. Cooks, J. Am. Soc. Mass Spectrom. 21, 572–579 (2010).
(14) H. Chen and Z. Miao, US20100059674.
(15) S. Zhang, Y.S. Shin, R. Mayer, and E. Basile, J. Anal. Appl. Pyrol. 80, 353–359 (2007).
(16) J.P. Williams, G.R. Hilton, K. Thalassinos, A.T. Jackson, and J.H. Scrivens, Rapid Commun. Mass Spectrom. 21, 1693–1704 (2007).
(17) D.R. Ifa, L.M. Gumaelius, L.S. Eberlin, N.E. Manicke, and R.G. Cooks, Analyst 132, 461–467 (2007).
(18) M.S. Bereman, T.I. Williams, and D.C. Muddiman, Anal. Chem. 79, 8812–8815 (2007).
(19) G. Kaur-Atwal, D.J. Weston, P.S. Green, S. Crosland, P.L.R. Bonner, and C.S. Creaser, Rapid Commun. Mass Spectrom. 21, 1131–1138 (2007).
(20) D.R. Justes, N. Talaty, I. Cotte-Rodriguez, and R.G. Cooks, Chem. Commun. 21, 2142–2144 (2007).
(21) C.C. Mulligan, D.K. MacMillan, R.J. Noll, and R.G. Cooks, Rapid Commun. Mass Spectrom. 21, 3729–3736 (2007).
(22) L. Cott-Rodriguez, H. Hernandez-Soto, H. Chen, and R.G. Cooks, Anal. Chem. 80, 1512–1519 (2008).
(23) A.L. Dill, D.R. Ifa, N.E. Manicke, Z. Ouyang, and R.G. Cooks, J. Chromatogr. B 877, 2883–2889 (2009).
(24) C. Wu, D.R. Ifa, N.E. Manicke, and R.G. Cooks, Analyst 135, 28–32 (2010).
(25) L.S. Eberlin, D.R. Ifa, C. Wu, and R.G. Cooks, Angew. Chem. Int. Ed. 49, 873–876 (2010).
(26) J. PóV. Vidová, G. Kruppa, V. Kobliha, P. Novák, K. Lemr, T. Kotiaho, R. Kostiainen, V. Havlicek, and M. Volný Anal. Chem. 81, 8479–8487 (2009).
(27) L. Nyadong, S. Late, M.D. Green, A. Banga, and F.M. Fernández, J. Am. Soc. Mass Spectrom. 19, 380–388 (2008).
(28) G.A. Harris, L. Nyadong, and F.M. Fernandez, Analyst 133, 1297–1301 (2008).
(29) H. Chen, A. Venter, and R.G. Cooks, Chem. Commun . 2042–2044 (2006).
(30) W.S. Law, R. Wang, B. Hu, C. Berchtold, L. Meier, H. Chen, and R. Zenobi, Anal. Chem. 82, 4494–4500 (2010).
(31) L. Zhu, G. Gamez, H. Chen, K. Chingina, and R. Zenobi, Chem. Commun. 559–561 (2009).
(32) H. Gu, H. Chen, Z. Pan, A.U. Jackson, N. Talaty, B. Xi, C. Kissinger, C. Duda, D. Mann, D. Raftery, and R.G. Cooks, Anal. Chem. 79, 89–97 (2007).
(33) K. Chingin, G. Gamez, H. Chen, L. Zhu, and R. Zenobi, Rapid Commun. Mass Spectrom. 22, 2009–2014 (2008).
(34) P. Martínez-Lozanoa and J. Fernández de la Mora, Int. J. Mass Spectrom. 265, 68–72 (2007).
(35) L. Zhu, G. Gamez, H.W. Chen, H.X. Huang, K. Chingin, and R. Zenobi. Rapid Commun. Mass Spectrom. 22, 2993–2998 (2008).
(36) B.J. McCullough, T. Bristow, G. O'Connor, and C. Hopley, Metabolomics 3, 101–104 (2007).
(37) H. Chen and R. Zenobi, Nat. Protoc. 3, 1467–1475 (2008).
(38) Z. Wu, K. Chingin, and H. Chen, Anal. Bioanal. Chem. 397, 1549–1556 (2010).
(39) H. Chen, S. Yang, A. Wortmann, and R. Zenobi, Angew. Chem. Int. Ed. 46, 7591–7594 (2007).
(40) R.B. Cody, J.A. Laramee, and H.D. Durst, Anal. Chem. 77, 2297–2302 (2005).
(41) Z. Takáts, I. Cotte-Rodriguez, N. Talaty, H. Chen, and R.G. Cooks, Chem. Commun. 1950–1952 (2005).
(42) F.J. Andrade, J.T. Shelley, W.C. Wetzel, M.R. Webb, G. Gamez, S.J. Ray, and G.M. Hieftje, Anal. Chem. 80, 2646–2653 (2008).
(43) J.D. Harper, N.A. Charipar, C.C. Mulligan, X. Zhang, R.G. Cooks, and Z. Ouyang, Anal. Chem . 80, 9097–9104 (2008).
(44) N. Na, M. Zhao, S. Zhang, C. Yang, and X. Zhang, J. Am. Soc. Mass Spectrom. 18, 1859–1862 (2007).
(45) J.M. Symonds, A.S. Galhena, F.M. Fernandez, and T.M. Orlando, Anal. Chem. 82, 621–627 (2010).
(46) L. Nyadong, G.A. Harris, S. Balayssac, A.S. Galhena, M. Malet-Martino, R. Martino, R.M, Parry, M.D. Wang, F.M. Fernandez, and V. Gilard, Anal. Chem. 81, 4803–4812 (2009).
(47) C. Petucci, J. Diffendal, D. Kaufman, B. Mekonnen, G. Terefenko, and B. Musselman, Anal. Chem. 79, 5064–5070 (2007).
(48) N.J. Smith, M.A. Domin, and L.T. Scott, Org. Lett. 10, 3493–3496 (2008).
(49) M. Zhou, J.F. McDonald, and Facundo M. Fernándeza, J. Am. Soc. Mass Spectrom. 21, 68–75 (2010).
(50) A.T. Navare, J.G. Mayoral, M. Nouzova, F.G. Noriega, and F.M. Fernández, Anal. Bioanal. Chem . 398, 3005–3013 (2010).
(51) M.A. Domin, B.D. Steinberg, J.M. Quimby, N.J. Smith, A.K. Greene, and L.T. Scott, Analyst 135, 647–658 (2010).
(52) G. Morlock and Y. Ueda, J. Chromatogr. A 1143, 243–251(2007).
(53) W. Eberherr, W. Buchberger, R. Hertsens, and C.W. Klampfl, Anal. Chem. 82, 5792–5796 (2010).
(54) H. Chen, J. Zheng, X. Zhang, M. Luo, Z. Wang, and X. Qiao, J. Mass Spectrom. 42, 1045–1056 (2007).
(55) H. Chen, B. Hu, and X. Zhang, Chin. J. Anal. Chem. 38, 1069–1088 (2010).
(56) Y. Zhang, Z. Yuan, H.D. Dewald, and H. Chen, Chem. Commun. 47, 4171–4173 (2011).
(57) G.J.V. Berkel, M.J. Ford, and M.A. Deibel, Anal. Chem. 77, 1207–1215 (2005).
(58) G.J.V. Berkel and V. Kertesz, Anal. Chem. 78, 4938–4944 (2006).
(59) J.H. Kennedy and J.M. Wiseman. Rapid Commun. Mass Spectrom. 24, 1305–1311 (2010).
(60) J. Dénes, M. Katona, A.d. Hosszú, N. Czuczy, and Z. Takáts, Anal. Chem. 81, 1669–1675 (2009).
(61) J. Li, H.D. Dewald, and H. Chen, Anal. Chem. 81, 9716–9722 (2009).
(62) Y. Zhang, Z. Yuan, H.D. Dewald, and H. Chen, Chem. Commun. 47, 4171–4173 (2011).
(63) R. Haddad, R. Sparrapan, and M.N. Eberlin, Rapid Commun. Mass Spectrom. 20, 2901–2905 (2006).
(64) R. Haddad, R. Sparrapan, and M.N. Eberlin, Anal. Chem. 80, 898–903 (2008).
(65) R. Haddad, H.S. Milagre, R.R. Catharino, and M.N. Eberlin, Anal. Chem . 80, 2744–2750 (2008).
(66) F. Huang and K.K. Murray, Rapid Commun. Mass Spectrom. 24, 2799–2804 (2010).
(67) M. Li, B. Hu, J. Li, R. Chen, X. Zhang, and H.W. Chen, Anal. Chem. 81, 7724–7731 (2009).
(68) I.F. Shieh, C.Y. Lee, and J. Shiea, J. Proteome Res. 4, 606–612 (2005).
(69) S.H. Yang, A.B. Wijeratne, L. Li, B.L. Edwards, and K.A. Schug, Anal. Chem. 83, 643–647 (2011).
(70) T.J. Kauppila, N. Talaty, P.K. Salo, T. Kotiaho, R. Kostiainen, and R.G. Cooks, Rapid Commun. Mass Spectrom. 20, 2143–2150 (2006).
(71) G.J.V. Berkel, B.A. Tomkins, and V. Kertesz, Anal. Chem. 79, 2778–2789 (2007).
(72) G. Paglia, D.R. Ifa, C. Wu, G. Corso, and R.G. Cooks, Anal Chem. 82, 1744–1750 (2010).
(73) S.P. Pasilis, V. Kertesz, G.J. Van Berkel, M. Schulz, and S. Schorcht, Anal. Bioanal. Chem. 391, 317–324 (2008).
(74) X. Sun, Z. Miao, Z. Yuan, P.B. Harrington, J. Colla, and H. Chen, Int. J. Mass Spectrom. 301, 102–108 (2011).
(75) G.K. Barbula, S. Safi, K. Chingin, R.H. Perry, and R.N. Zare, Anal. Chem. 83, 1955–1959 (2011).
(76) X. Ma, M. Zhao, Z. Lin, S. Zhang, C. Yang, and X. Zhang, Anal. Chem. 80, 6131–6136 (2008).
(77) M.C. Jecklin, S. Schmid, P.L. Urban, A. Amantonico, and R. Zenobi, Electrophoresis 31, 3597–3607 (2010).

![Huck_Fig 2-[41825010]-{584590}_t-746305-1408610899253.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2Ff6261c79607ac53f41c790ed011e5b2e0d4f0626-200x140.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Huck_Fig 2-[41825010]-{584590}_t-746305-1408610899253.gif")
![Roy figure 5-[41827930]-{585410}-746304-1416911904998.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F5e8a7a8b42be4c1287e42a9e19c61a898804cfe5-700x592.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Roy figure 5-[41827930]-{585410}-746304-1416911904998.gif")
![Schug_Fig 3-[41827280]-{641210}_t-746310-1408610889870.jpg](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F6da0479f1e71c57976064cc832407ebe8e770158-200x104.jpg%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Schug_Fig 3-[41827280]-{641210}_t-746310-1408610889870.jpg")
![Heinle Figure 1-[41825860]-{582380}_t-746308-1408610893731.gif](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fchroma%2F309898def0ba4426d4cd28df126a523915eb3959-200x103.gif%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Heinle Figure 1-[41825860]-{582380}_t-746308-1408610893731.gif")






. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.



