LC Trouble Everywhere
LCGC North America
This month, we look at calculating injection volume, choosing a wash solvent, and handling doubts about reference standard purity.
No matter where liquid chromatography is used, it is never immune to problems.
One of the privileges of teaching liquid chromatography (LC) classes for a living is that I get to meet chromatographers from around the world during my travels. As I write this installment, I'm just finishing six weeks of classes, where I have had a chance to discuss problems that attendees were having with a variety of LC applications. Although the topics in this column originate in Poland, Turkey, and Argentina, they are no different than those that arise in Singapore, Galway, Charlottetown, or Huntsville. Let's look at a couple of problems that students had related to both conventional LC and ultrahigh-pressure LC (UHPLC). These problems apply equally to both techniques.
How Much Can I Inject?
One of my students from Poland, who was just starting out using UHPLC, asked if she could inject 5 µL of sample onto her UHPLC column without problems. Her conditions comprised a 75 mm × 2.1 mm column packed with 1.8-µm diameter particles operated at a flow rate of 0.8 mL/min. It is fairly easy to perform a few simple calculations and answer her question.
A good rule of thumb (1) is that you can inject ≈15% of the volume of the first peak of interest without causing a more than ≈5% increase in peak broadening, as long as you use mobile phase for the injection solvent. We need to determine the volume of the first peak of interest to see how this applies in the present case. We will assume a retention factor, k, of 1 for the first peak; you can make similar calculations for other retention factors.
First, we need to estimate a column plate number (efficiency), N, which can be done as
N = 300 L/dp [1]
where L is the column length in millimeters and dp is the particle diameter in micrometers. A well-packed column tested with an ideal solute will give larger values of N, but equation 1 is a reasonable estimate for real samples. With our L = 75 mm, dp = 1.8 µm column, we get N = 12,500.
Next, it is necessary to estimate the column volume, VM, in milliliters, using the equation
VM ≈ 5 × 10-4Ldc2 [2]
where dc is the column diameter in millimeters. For the 75 mm × 2.1 mm column we get 0.165 mL. From this we can get the column dead time, t0, by dividing by the flow rate, 0.8 mL/min: 0.21 min. (Note: If you are trying to repeat these calculations, carry along several extra decimal places of precision — I'm rounding the numbers for simplicity of presentation).
With the knowledge of the column dead time, we can calculate the retention time, tR, for our k = 1 peak by rearranging the equation for k:
tR = t0 (1 + k) [3]
This gives tR = 0.41 min.
The next to last step is to estimate the peak volume of the first peak. We estimated N with equation 1, but if we were measuring N from an isocratic chromatogram, we would use
N = 16 (tR/w)2 [4]
where w is the baseline width of the peak measured between tangents drawn along the sides of the peak. Solving equation 4 for the peak width, we get
w = 4 tR/N0.5 [5]
For tR = 0.41 min and N = 12,500, this gives w = 0.015 min, or at 0.8 mL/min, w = 11.8 µL.
Using our 15% rule of thumb from the beginning of our discussion, an injection of (0.15 × 11.8 µL) = 1.8 µL, or ≈ 2 µL would be allowed in the mobile phase. This is less than half the desired 5-µL injection, so it is expected that excess peak broadening would be observed with a 5-µL injection. Armed with this knowledge, I would perform a series of injections of 1, 2, 3, 4, and 5 µL and observe the effect on the peak width of the first peak of interest. I agree wholeheartedly with Izaak Kolthoff, the father of analytical chemistry, who is credited with the quote, "Theory guides, experiment decides." So, use the results of the injection-volume experiment for the final decision.
Alternatives
The above example illustrates one of the challenges of UHPLC — reducing the sample volume sufficiently to avoid band broadening. Perhaps this could be accomplished if any preceding sample-preparation steps involved reconstitution of a dried extract. For example, if an evaporation-to-dryness step was followed by reconstitution in 50 µL of mobile phase, this volume could be reduced to 10 µL and now a 1-µL injection would contain the same mass of sample as 5 µL of the 50-µL volume.
Because the band-broadening effect is worse for the first peak in the chromatogram, it may be possible to mitigate the problem by increasing the retention of the first peak. If you repeat the above calculations with several different k-values for the first peak, you'll see that a 5-µL injection would be allowed with k = 4–5. This would likely be too long to wait (≈1 min) for the first peak for most impatient chromatographers — after all, you bought that UHPLC system so you could have short run times!
A third option to solve this problem might be to use on-column concentration. When the injection solvent is more than 20% weaker than the mobile phase, it is possible to inject large volumes of sample because the weak solvent causes the sample to accumulate at the head of the column until the injection solvent washes through. For example, if the mobile phase was 65% acetonitrile and 35% buffer, injection in ≤45% acetonitrile would be expected to allow injection volumes much larger than the 2-µL injection calculated above. Thus, if the desired injection of 5 µL was in mobile phase, you could dilute the sample twofold with buffer and inject 20 µL of the sample (now in ≈33% acetonitrile) and put the desired sample mass on the column with less chance of unwanted band broadening.
As with the conclusion of the previous section, try one or more of these alternative solutions and see what works.
Wash Solvent Choice
A student from Buenos Aires was using LC for the analysis of a drug product. He was having problems with carryover, so he was experimenting with different autosampler wash solvents to see if he could mitigate the problem. With some wash solvents he noticed significant peak broadening for his sample, whereas others did not cause problems. Unfortunately, when he used the most effective wash solvent, 100% methanol containing 0.1% formic acid, he observed the worst peak shape. He wondered how he could select a wash solvent that would reduce carryover, yet maintain good peak shape.
One of the most popular autosamplers in use today is the needle-in-loop design. This configuration typically uses a 100-µL sample loop that has the injection needle as a part of the loop. To inject, the needle is connected to the LC mobile-phase flow path with a high-pressure seal. The nice thing about this design is that, because the needle is in the flow path, 100% of the sample (up to the loop volume) is injected. However, the entire loop contents of 100 µL are injected every time, even with a small sample injection. For example, if a 1-µL injection is made, the loop contains 1 µL of sample plus 99 µL of solvent. Usually the loop is backflushed onto the column so that the sample goes onto the column first, followed by the remainder of the loop contents. If the loop is filled with mobile phase (or a weaker solvent), there is no problem; it is as if a 1-µL sample loop was used. However, if the solvent in the loop is stronger than the mobile phase, the injection plug can be distorted and cause band broadening. Obviously, it is desirable to avoid injecting an excess of strong solvent so as to avoid unwanted band broadening.
So, how do we avoid injecting too much of a strong solvent? One way is to perform the wash cycle while the sample loop is in the flow path. With most LC systems, when the injection is made, the injection valve stays in the inject position during the run. If the autosampler wash cycle is performed while the injection valve is in the inject position, the remainder of the autosampler is washed with the wash solvent, but the sample loop is not. The sample loop is thoroughly flushed with mobile phase during the run, which is usually sufficient to clean it before the next injection — after all, the sample is soluble in the mobile phase, so the mobile phase should be good enough to flush the loop. If, however, the loop is washed in the load position, the wash solvent will remain in the loop when the next sample is aspirated. Thus, any remaining wash solvent will get injected with the sample. If this wash solvent is too strong, peak distortion is likely. Some autosamplers provide for the use of two wash solvents. If this is the case, the solution to the problem is simple. First, wash with the strong wash solvent in the load position to remove any residues from the sample loop, then wash with mobile phase (or a weaker solvent). Now when the sample is picked up, the remainder of the loop will be filled with a non-band-broadening solvent, so peak distortion should not occur. A final option would be to use a smaller-volume sample loop, if this is an option with your autosampler. Using a filled-loop injection with a 1-µL loop volume, for example, would allow injection of 1 µL of sample and because none of the wash solvent would remain in the loop, the choice of wash solvent composition would be of little concern.
Reference Standard Purity
In Turkey, a student complained that he had purchased a reference standard that was claimed to be >99% pure in the certificate of analysis, but when he ran it on his LC system, he found impurity peaks that greatly exceeded the <1% claim. He showed me a chromatogram that he obtained with a large analyte peak and a smaller impurity peak that indeed was more than 1% of the reference standard. He also had a copy of the certificate of analysis that contained a chromatogram showing a single peak that looked similar to the one I've recreated in Figure 1. The mobile phase was 90% acetonitrile, 10% water on a C18 column for the certificate of analysis; unfortunately, I did not note the conditions for his analysis.


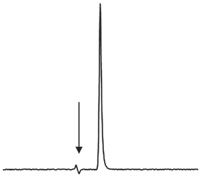
Figure 1: Reconstructed chromatogram for a certificate of analysis. The arrow marks the column dead time.
However, even with this limited amount of information, it is possible to make an educated guess about the source of the problem. If you recall from previous installments of this column, I've stated many times that the ideal isocratic chromatogram will have peaks in the 2 < k < 10 range, or if this is not possible, 1 < k < 20. This gives the analytes sufficient time to interact with the column to achieve "good" chromatography, yet keeps the run time from being excessive. Normally, we calculate the retention factor, k as
k = (tR – t0)/t0 [6]
where tR and t0 are the retention time and column dead time as noted in equation 3. I did not include a time axis in Figure 1, because I can't remember what it was in the certificate of analysis, but the units in equation 6 cancel, so we can use a ruler to calculate k from the chromatogram. The first disturbance in the baseline (arrow in Figure 1) marks t0 and we can use the top of the peak as tR to calculate k ≈ 0.3. This is much less than the desired minimum k = 2. When peaks are eluted with k << 1, there is little time for the analyte to interact with the column, and the chances of having unresolved peaks present is increased. This is especially important when using a chromatogram to certify peak purity. The small tail on the peak may be peak tailing that is normal with many peaks or it may be a subtle indication that an impurity is present. There is no excuse for producing a chromatogram like this for a certificate of analysis, except laziness, ignorance, or impatience. It would be easy to increase k for the peak from 0.3 to 2 < k < 10 by changing to a weaker mobile phase, such as 70% acetonitrile, 30% water. At that point, a more convincing case could be made to show that the peak was indeed >99% pure. Although I don't remember the conditions of the user's chromatogram, I do remember that the analyte peak was significantly broader than that in the certificate of analysis, suggesting that the retention time was much larger. My answer to the question was that the certificate of analysis didn't convince me of the purity of the reference standard, so I would be more likely to trust the analyst's chromatogram in which k >> 0.3.
Conclusions
It doesn't matter where we live; LC problems have no international boundaries. In fact, I've noticed that chromatographic terminology often is adopted directly into many different languages. In some ways, chromatography really is a universal language. It reminds me of a time when a visitor from one of the Asian countries visited my son's classroom when my son was about 12 years old. The visitor asked if the kids wanted to learn some of his language. All were excited about the prospect. The visitor carefully recited, "computer," "Coca-Cola," and "McDonalds." Maybe he should have included "chromatography."
John W. Dolan "LC Troubleshooting" Editor John Dolan has been writing "LC Troubleshooting" for LCGC for more than 25 years. One of the industry's most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources in Walnut Creek, California. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com.


John W. Dolan
References
L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography, 3rd Ed. (Wiley, Hoboken, New Jersey, 2010), p. 120.


 Applications in Biotechnology")













Understanding FDA Recommendations for N-Nitrosamine Impurity Levels
April 17th 2025We spoke with Josh Hoerner, general manager of Purisys, which specializes in a small volume custom synthesis and specialized controlled substance manufacturing, to gain his perspective on FDA’s recommendations for acceptable intake limits for N-nitrosamine impurities.



