Introduction to SPME for In Vivo Analysis
LCGC Europe
The use of in vivo SPME offers important advantages over conventional methods according to the authors, including simplified sample clean-up, fast stabilization of unstable analytes, elimination of enzymatic degradation after extraction and reduced ion suppression for mass spectrometry analyses.
Solid-phase microextraction (SPME) was originally introduced as a solvent-free sample preparation technique ideally suited for analysis of fragrance compounds and environmental pollutants requiring gas chromatographic (GC) analysis. Introduction by thermal desorption into a standard GC injector helped to make this technique widely used. Its applicability for additional compound classes including pesticides, drugs and endogenous chemicals requiring liquid chromatographic (LC) analysis was demonstrated later, where the desorption of extracted analyte into a minimal volume of solvent compatible for injection into LC instrumentation was employed. Recently, new SPME sorbent phases have been introduced to further broaden the range of applications of the technology, and facilitate both on-site and in vivo sampling and sample preparation from dynamic living systems. This article briefly introduces the theory relevant to such applications and provides examples of its use.
Solid-phase microextraction (SPME) is an extraction and pre-concentration technology developed to address the need for rapid sample preparation procedures both in the laboratory and in the field (1,2). In the past two decades it has found widespread use as a sample preparation tool for investigations using gas chromatography (GC) and liquid chromatography (LC). As a result of its simple design and operation, it has also enabled automation and highthroughput sample preparation for both types of separation.



The basic concept of the technology in its current commercial configuration is a sorbent-coated rod that is put into contact with a sample (gaseous, liquid, semi-solid) or the headspace of liquids or solids. The sorbent is selected to have a good affinity with the analyte of interest in the sample. After a pre-defined exposure time, sufficient analyte will have moved from the sample to the sorbent to permit quantitative analysis. The amount extracted is proportional to the original concentration of analyte in the sample, which permits the easy determination of sample concentration (3).
Although up till now SPME devices have been used mainly in laboratory applications, recent research has been directed towards field monitoring, particularly for clinical, environmental and industrial hygiene applications. A new line of biocompatible SPME devices has been specifically developed to better exploit these opportunities, particularly for in vivo analysis (4). These devices use a C18 porous silica sorbent (similar to particles typically used in HPLC columns or as SPE sorbents) in a proprietary biocompatible binder [45 µm thickness, 15 mm length of the coating] (Supelco, Bellefonte, Pennsylvania, USA).
The binder used is a nonswelling polymer which resists fouling by biological matrix components (5). The solid support used for these probes is a flexible metal alloy (0.008" dia), and the coatings are housed inside a 22-gauge hypodermic needle with a seal incorporated to prevent sample loss during sampling. Because of the C18 extraction phase, they perform absorptive extraction.
The devices may be employed for single use and are ideally suited for application, either in vitro to sample directly from whole blood or plasma in sample vials sealed with hole caps and septa, or through an injection bulb on an intravenous catheter for in vivo studies. Devices without the attached hypodermic needle are also available where sealing is not critical (for example, tissue sampling or sampling from open vials). The operating principles of such devices are analogous to those of the original devices. The modifications are made for greater convenience in given applications.
An important advantage of SPME, particularly for on-site sampling, is the possibility of performing analyses without pre-defining a specific sample size. The reason for this may be understood from a brief review of SPME theory. The amount of an analyte extracted from a sample by SPME at equilibrium is given in Equation 1 (6):

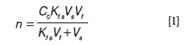
where C0 is the initial sample concentration of the analyte, n is the amount of analyte extracted by the device, Vs is the sample volume, Vf is the fibre sorbent volume and Kfs is the analyte distribution constant between the sorbent and sample matrix. However, when sample size is large relative to the fibre capacity, the amount extracted is insignificant relative to the amount present in the sample (that is, conditions of negligible depletion). Specifically this occurs when Vs >> VfKfs. In this case Equation 1 reduces to Equation 2. This renders the amount of analyte extracted by SPME independent of the sample volume.

This allows for the application of SPME to on-site analysis and eliminates the need to remove a representative sample from the system under study to perform the analysis. From a bioanalytical perspective, this permits the use of SPME to directly sample blood or tissue of animals in vivo, without having to withdraw an appropriate biofluid/tissue sample. Several applications for in vivo SPME analysis where this benefit has been exploited in animals have appeared in recent years.
A determination of the magnitude of sample volume required to satisfy this condition may be made from calibration data using Equation 1. Where mass extracted is plotted against sample concentration, the fibre constant (VfKfs) may be derived from the slope of the resulting linear regression curve.
Selection of the extraction time should also be given some consideration. While full equilibrium is practical for gas sample extractions, where diffusion coefficients in the sample are high, it is commonly impractical for extractions from liquid samples. This is particularly true for in vivo analysis, where a very short (pre-equilibrium) extraction time is usually preferred. Very short extraction times typically fall in the kinetic region of the adsorption time uptake profile.
This is beneficial for in vivo extraction calibration because in this region uptake is controlled by diffusion across the boundary layer rather than the kinetics and/or thermodynamics of sorbent interactions. In such cases, calibration of mass uptake relative to sample concentration may be performed by monitoring the loss of an internal standard pre-loaded onto the sorbent.
Where uptake kinetics are controlled by the boundary layer diffusion, there is a symmetry between analyte uptake and standard loss. Although it is almost always valid, it is prudent to verify that the relationship holds for a particular experiment. In our experience, this symmetry only breaks down in a few rare cases where the kinetics of analyte interaction with the sorbent surface are very slow and the boundary layer very thin. In the majority of cases, loss of pre-loaded internal standard from the sorbent may be used to control for variable extraction time and sample convection conditions, as well as providing for calibration of sample concentration.
Applications for In Vivo Analysis
Dogs: Initially, in vivo SPME studies were applied to study the pharmacokinetics (PK) of various drugs in directly from the veins of animals (7–12). Specifically SPME devices based on polypyrrole and polyethylene glycol coatings were used for direct extraction of diazepam and its metabolites form the flowing blood of beagle dogs (13). LC coupled to tandem mass spectrometry (LC–MS–MS) was used for analysis. Two calibration strategies, external and kinetic calibration, were used to correlate the extracted amount of analytes with the in vivo concentrations. The probes were exposed to the flowing blood for 2 min for equilibrium extraction and 30 s for kinetic calibration. The fast microextraction technique caused minimal disturbances to the drug/protein binding equilibria so additional information on the free concentration could be obtained. These studies exhibited the quantitative capability of SPME for in vivo sampling.
Rodents: Although rats are extensively used for traditional PK studies in drug discovery, there is an increased interest in the use of mice because of the availability of various strains of gene knockout mice and indications that mouse data may be more useful than rat data for allometric scaling of PK parameters from animals to humans (9). Traditional PK studies in mice rely on the sacrifice of multiple animals at each time point of interest, which introduces interanimal variation and dosing inaccuracies into the PK data. This disadvantage has been addressed by performing studies where all samples are acquired from a single animal by serial blood sample withdrawal, but to date such studies have been difficult to execute in mice because a limited circulating blood volume (< 2 mL/25 g mouse) and difficulties in the handling and sample preparation of the small blood sample volumes.
A specialized sampling interface was developed to permit monitoring of small rodents (mice and rats) (11, 12, 14) and the blood draw-free SPME technique was applied for in vivo pharmacokinetic studies drugs directly in the bloodstream (8). The SPME probes were inserted into the surgicallyimplanted interface through which blood was allowed to circulate. The main advantages of single rodent PK studies include: (1) a reduction in animal use which is particularly important when using rare or expensive strains of mice; (2) a reduction of dosing errors in the data; (3) a decrease in the amount of compound needed to perform the study, which is particularly advantageous in early drug discovery where the quantity of test compound is limited; (4) elimination of the effects of anesthesia from the PK data; (5) the ability to study interanimal variation; and (6) the opportunity to perform multiple and/or simultaneous pharmacokinetics/pharmacodynamics investigations in the same mice (10–12, 14). The PK data obtained from serial SPME sampling was equivalent to the data obtained using terminal sampling from multiple animals, indicating the validity of this approach (12).
Brain: An in vivo SPME technique with conventional SPME fibres was successfully used for the detection of toluene levels in the brains of conscious, free-moving mice (15, 16). In order to measure the pharmacokinetics of inhaled toluene in the brains of mice, a SPME fibre was inserted into the hippocampus (CA1) through a cannula fixed onto the animal. BALB/c mice were exposed to toluene for 30 min. The pharmacokinetics of toluene in the brain of mice exposed to 50 ppm toluene showed that the toluene level decreased rapidly after the exposure, and returned to control levels after 60 min. The technique does not require lethal animal testing, and has the potential to become a useful tool for studies on the field of neurotoxicology (16).
We have also recently studied the use of the new SPME fibres for monitoring brain function (17). Probes with a short (4 mm) sorbent length were prepared and used to monitor selected neurotransmitter levels in response to electrical stimulation of the brain. The SPME probes were positioned in the target brain region (hippocampus) through a surgically implanted microdialysis cannula, similar to the positioning of a microdialysis probe. The neurotransmitters studied were: serotonin, dopamine, γ-aminobutyric acid and glutamic acid and these were detected by LC–MS–MS. No significant changes were observed for the studied neurotransmitters before and during electrical stimulation, except for serotonin, which showed an approxiamte 2–3× (~ 50 pg/mL–400 pg/mL) increase during deep brain stimulation (DBS). Relative standard deviations (RSD) for serotonin at basal concentrations ranged from 3–20%. The results were equivalent in the target brain region in both the left and right hemispheres of all four rats studied. The SPME method addresses some of the limitations of microdialysis for these analyses, such as better sensitivity for a given sampling time, the unrestricted movement of the animals during sampling as a result of the lack of peripheral tubing, as well as the elimination of the significant signal suppression as a result of matrix components of the dialysate in microdialysis.
The increase in serotonin after DBS in the present study was similar in magnitude to previous results obtained by Hamani et al. using microdialysis (18). This suggests that SPME as a tissue sampling tool may be a valid alternative approach to measure neurotransmitter changes after particular interventions.
Fish: Over the past five years, we have applied SPME to the in vivo determination of pharmaceuticals in fish tissue using direct muscle sampling (19–22). A significant advantage of SPME fibres is their ability to extract a variety of trace contaminants from fish tissues without lethal sampling. This means it is possible to monitor the presence of a target analyte in the same fish over several days. We have also evaluated the use of the devices for analysis of fish fillets after lethal sampling. In either case, sampling involves using a hypodermic needle to create a small track in the tissue followed by inserting the SPME sampler so that the sorbent is in direct contact with the target tissue for a few minutes.
This is followed by withdrawing the needle and measuring the chemicals removed by the sorbent after transport to the laboratory. By pre-loading internal standard onto the fibre we are able to determine the concentrations of the chemicals in the tissue based on the loss of standard from the fibre during sampling. We have demonstrated repeatedly that the results obtained have the same accuracy as other more conventional methods of analysis (12, 7, 23). The system is much simpler to use than conventional analysis, thus field technicians do not require advanced training in chemical analysis. Laboratorybased steps are also greatly simplified. The method has been demonstrated for the analysis of pharmaceuticals to low to moderate ng/g levels in greenside darter, fathead minnow and rainbow trout (24). We have also demonstrated a method to calculate bioaccumulation factors for different species and pollutants (19, 20, 25).
Metabolomics: Currently there is an increased interest in the development of global or untargeted metabolomic profiling methods which can capture hundreds or thousands of metabolites in a single liquid chromatography–mass spectrometry (LC–MS) run. The choice of sampling, sample collection and sample preparation strategies plays an important role in the quality of metabolomics data during these studies (26). Issues with incomplete metabolism quenching, ionization suppression and metabolite instability have been well-documented and can adversely impact data interpretation. In vivo solidphase microextraction (SPME) is an attractive sampling method for global metabolomics studies by LC–MS.
The potential benefits of SPME for metabolomics studies, such as the capture of short-lived and unstable metabolites and good sample throughput, have been demonstrated for the detection of biotransformation intermediates and both global and targeted metabolomics studies using GC–MS primarily in headspace mode (27–33). However, to date SPME in direct extraction mode in combination with LC–MS analysis has been used in the main for the analysis of a small number of a priori selected target compounds (23, 34). The main difficulty with applying these existing workflows to envisioned global metabolomics studies in complex biological matrices, such as blood or tissue, is the lack of coatings that can simultaneously extract both polar and nonpolar species using direct extraction mode. In fact, only four SPME coatings have been available commercially until now: poly(dimethylsiloxane) (PDMS), poly(dimethylsiloxane/divinylbenzene) (PDMS/DVB), polyacrylate (PA) and Carbowax-templated resin (CW-TPR). They exhibit poor extraction efficiency toward polar analytes, can suffer from the fouling of the extraction phase upon exposure to biological samples and are too expensive, which makes the use of multiple fibres during large-scale metabolomics studies cost prohibitive.
We have recently reported on the use of SPME in direct extraction mode for untargeted metabolite profiling studies in combination with LC–MS using the new generation biocompatible SPME probes (35). We have shown that mixed-mode, phenylboronic acid and polystyrenedivinylbenzene coatings are particularly suitable for this type of application. We have demonstrated for the first time that it is possible to extract hundreds of endogenous metabolites with a single coated fibre, whereas previously all existing reports of direct immersion SPME deal with targeted analysis of a few a priori selected analytes.
For in vitro studies on human biological fluids, SPME can serve as a complementary technology to the widely used solvent precipitation approaches. It has been shown to provide improved quantitative information on polar species because of a reduction in ionization suppression, complementary metabolite coverage and information on the biologically relevant free concentration. In contrast to ultrafiltration, SPME reduces ionization suppression and improves coverage of hydrophobic species. These are typically highly bound and may not be observed by ultrafiltration either as a result of adsorption to the membrane or a high degree of protein binding. Further work is currently underway to exploit the in vivo sampling capability of SPME and demonstrate that SPME can more accurately capture the metabolome at the time of sampling, in particular for the unstable metabolites and/or metabolites with fast turnover rates.
Conclusions
The use of in vivo SPME offers important advantages over conventional methods, such as simplified sample cleanup, fast stabilization of unstable analytes, elimination of enzymatic degradation after extraction and reduced ion suppression for mass spectrometry analyses. Furthermore, because both sampling and sample clean-up are combined into one step, the number of sample preparation steps, which can potentially result in loss of analyte or accidental contamination, is minimized (9). A recent article in Nature Protocols details all of the steps involved in performing in vivo SPME for intravenous drug and metabolite monitoring (36).
Heather Lord is a research associate in the group of Professor Pawliszyn with experience in analytical device and method development, sorbent design and solid-phase sample preparation. She has authored over 40 scientific papers in biochemical toxicology and bioanalytical method development. Her primary research interests are in the areas of in vivo sample preparation and new bioanalytical methods development.
The primary focus of Professor Pawliszyn's research programme is the design of highly automated and integrated instrumentation for the isolation of analytes from complex matrices and the subsequent separation, identification and determination of these species. Currently, his research is focused on the elimination of organic solvents from the sample preparation step to facilitate on-site monitoring and in vivo analysis. He is also exploring the application of computational and modelling techniques to enhance performance of sample preparation, as well as the development and application of imaging detection combined with microseparation approaches.
References
(1) J. Pawliszyn and S. Liu, Anal. Chem., 59(10), 1475–1478 (1987).
(2) R.G. Belardi and J. Pawliszyn, Water Qual. Res. J. Can., 24(1), 179–191 (1989).
(3) C.L. Arthur and J. Pawliszyn, Anal. Chem., 62(19), 2145–2148 (1990).
(4) D. Vuckovic, R. Shirey, Y. Chen, L. Sidisky, C. Aurand, K. Stenerson and J. Pawliszyn, Anal. Chim. Acta, 638(2), 175–185 (2009).
(5) L. Musteata, M. Musteata and J. Pawliszyn, Anal. Chem., 79(18), 6903–6911 (2007).
(6) J. Pawliszyn, Solid phase microextraction, theory and practice, J. Pawliszyn, Ed.; Wiley-WCH, Inc.: USA (1997).
(7) H.L. Lord, R.P. Grant, M. Walles, B. Incledon, B. Fahie and J.B. Pawliszyn, Anal. Chem., 75(19), 5103–5115 (2003).
(8) F.M. Musteata, M.L. Musteata and J. Pawliszyn, Clin. Chem., 52(4), 708–715 (2006).
(9) X. Zhang, A. Es-Haghi, F.M. Musteata, G. Ouyang and J. Pawliszyn, Anal. Chem., 79(12), 4507–4513 (2007).
(10) A. Es-haghi, X. Zhang, F.M. Musteata, H. Bagheri and J. Pawliszyn, Analyst, 132(7), 672–678 (2007).
(11) F.M. Musteata, I. de Lannoy, B. Gien and J. Pawliszyn, J. Pharm. Biomed. Anal., 47(4–5), 907–912 (2008).
(12) D. Vuckovic, I. de Lannoy, B. Gien, Y. Yang, M.F. Musteata and J. Pawliszyn, J. Chromatogr. A, 1218(21), 3367–3375 (2011).
(13) F.M. Musteata, M.L. Musteata, J. Pawliszyn, Clin. Chem., 52(4), 708–715 (2006).
(14) D. Vuckovic, X. Zhang, E. Cudjoe and J. Pawliszyn, J Chromatogr. A, 1217(25), 4041–4060 (2010).
(15) T.T. Win-Shwe, D. Mitsushima, D. Nakajima, S. Ahmed, S. Yamamoto, S. Tsukahara, M. Kakeyama, S. Goto and H. Fujimaki, Toxicol. Lett., 168(1), 75–82 (2007).
(16) D. Nakajima, T.T. Win-Shwe, M. Kakeyama and H. Fujimaki and S. Goto, NeuroToxicology, 27(4), 615–618 (2006).
(17) E. Cudjoe, C. Hamani, E. Hoque, I. dé Lannoy, V. Saldivia, H. Sun and J. Pawliszyn, Solid phase microextraction: The potential for in vivo quantification of endogenous small polar molecules in biological matrices. Poster #2967, ASMS 59th Conference (Denver, CO, USA) (2011).
(18) C. Hamani, D. Mustansir, C.E. Macedo, M.L. Brandão, J. Shumake, F. Gonzalez-Lima, R. Raymond, A.M. Lozano, P.J. Fletcher and J.N. Nobrega, Biol. Psychiatry,67(2), 117–124 (2010).
(19) S.N. Zhou, K.D. Oakes, M.R. Servos and J. Pawliszyn, Environ. Sci. Tech., 42(16), 6073–6079 (2008).
(20) X. Zhang, J. Cai, K.D. Oakes, F. Breton and M.R. Servos and J. Pawliszyn, Anal. Chem., 81(17), 7349–7356 (2009).
(21) X. Zhang, K.D. Oakes, S. Cui, L. Bragg, M.R. Servos and J. Pawliszyn, Environ. Sci. Tech., 44(9), 3417–3422 (2010).
(22) S.N. Zhou, K.D. Oakes, M.R. Servos and J. Pawliszyn, Env. Sci. Technol., 42(16), 6073–6079 (2008).
(23) F. Musteata and J. Pawliszyn, J. Biochem. Biophys. Methods, 70(2), 181–193 (2007).
(24) S. Wang, K. Oakes, L. Bragg, J. Pawliszyn, G. Dixon and M.R. Servos, Chemosphere, 85(9), 1472–1480 (2011).
(25) G.K. Ouyang, G.K. Oakes, L. Bragg, S. Wang, S. Cui, M. Servos, D.G. Dixon and J. Pawliszyn, Environ. Sci. Technol., 45(18),
7792–7798 (2011).
(26) D. Ryan and K. Robards, Anal. Chem., 78(23), 7954–7958 (2006).
(27) J.C.R. Demyttenaere, R.M. Morina, N. De Kimpe, P. Sandra, J.Chromatogr. A, 1027(1–2), 147–154 (2004).
(28) A. Halasz and J. Hawari, J. Chromatogr. Sci., 44(7), 379–386 (2006).
(29) L.E. Hurd, F.R. Prete, T.H. Jones, T.B. Singh, J.E. Co and R.T. Portman, J. Chem. Ecol., 30(1), 155–166 (2004).
(30) D. Zimmermann, M. Hartmann, M.P. Moyer, J. Nolte and J.I. Baumbach, Metabolomics, 3(1), 13–17 (2007).
(31) H.A. Soini, K.E. Bruce, I. Klouckova, R.G. Brereton, D.J. Penn and M.V. Novotny, Anal. Chem., 78(20), 7161–7168 (2006).
(32) Y. Xu, S.J. Dixon, R.G. Brereton, H.A. Soini, M.V. Novotny, K. Trebesius, I. Bergmaier, E. Oberzaucher, K. Grammer and D.J. Penn, Metabolomics, 3(4), 427–437 (2007).
(33) S. Riazanskaia, G. Blackburn, M. Harker, D. Taylor and C.L.P. Thomas, Analyst, 133(8), 1020–1027 (2008).
(34) F.M. Musteata and J. Pawliszyn, TrAC, Trends Anal. Chem., 26(1), 36–45 (2007).
(35) D. Vuckovic and J. Pawliszyn, Anal. Chem., 83(6), 1944–1954 (2011).
(36) H.L. Lord, X. Zhang, F.M. Musteata, D. Vuckovic and J. Pawliszyn, Nature Protocols, 6(6), 896–924 (2011).

















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

