The Ideal Chromatography Data System for a Regulated Laboratory, Part I: The Compliant Analytical Process
LCGC North America
This is the first of a four part series looking at what functions and features the authors believe should be in a future chromatography data system (CDS) working within a regulated analytical laboratory. The first part of this series is aimed at setting the scene of where and how a CDS fits within the laboratory operation. In using the term CDS, in the future, this can refer to either a traditional CDS that is familiar to readers or the CDS functions could form part of another informatics solution such as Electronic Laboratory Notebook (ELN) or Laboratory information Management System (LIMS). In the next three parts we make 15 recommendations for improvements to the system architecture, the requirements for electronic working and regulatory compliance.
This article is the first of a four-part series looking at what functions and features the authors believe should exist in an ideal chromatography data system (CDS) of the future, designed for use in a regulated analytical laboratory. The first part of this series sets the scene of where and how a CDS fits into a laboratory operation. In the next three parts we make 15 recommendations for improvements to the system architecture, new CDS functions to enable fully electronic workflows, and features to ensure regulatory compliance.
Chromatography is a major analytical technique, especially in a regulated analytical laboratory, where chromatographic analyses can comprise up to 80% of the total analytical workload in some organizations. Automation of chromatographic analysis (instrument control, data acquisition, integration, and reporting of results) is undertaken by a chromatography data system (CDS). Unfortunately, CDS software has been at the heart of several recent data falsification cases (1), which demonstrates that we need a more systematic and structured approach to designing the ideal CDS, so that a CDS can be used not only to improve the speed and efficiency of the chromatographic process but also to ensure regulatory compliance. The regulations we refer to in this series of articles are the GXP regulations, a term that includes good laboratory practice (GLP), good manufacturing practice (GMP), and to a lesser extent good clinical practice (GCP).
It is important to understand that the current versions of CDS software were designed and released before the current regulatory focus on data integrity. Even if a CDS has the features required to enable chromatographers to carry out their work electronically, many laboratories use the system manually or as a hybrid (with signed paper printouts from the associated electronic records) and in many cases coupled with the use of a spreadsheet to undertake calculations that should really be performed in the CDS. This approach wastes the investment in the CDS and adds cost, risk, and complexity to the overall analytical process. Furthermore, many organizations do not know when and when not to perform manual integration. Manual integration was discussed in a recent “Questions of Quality” column (2).
The four-part series will focus mainly on the functionality required in CDS software. (And we will use the term CDS to refer to either a traditional CDS or to a future system in which current CDS functions could form part of another informatics solution such as an electronic laboratory notebook [ELN] or laboratory information management system [LIMS].) The further development of chromatographic instruments is relatively limited and moves forward incrementally, but there are significant advances that can still be made in CDS software applications that can result in major improvements to efficiency, effectiveness, and compliance within a regulated laboratory.
Chromatography data systems automate a variety of chromatographic processes that vary from conventional high performance liquid chromatography (HPLC) and ultrahigh-pressure LC (UHPLC), conventional and capillary gas chromatography (GC), and also HPLC and GC coupled with a range of mass spectrometry (MS) detectors. Many chromatography-MS systems have CDS applications that have come from a research environment into the regulated environment, and these data systems are ill-prepared for use in a regulated laboratory because they have system architectures and features that do not necessarily ensure data integrity. The scope of this series of articles includes these chromatography data systems. The principles outlined in these articles should also be applicable to laboratories working under other quality systems such as International Organization for Standardization (ISO) 17025. Also, although this article series focuses on CDS software, the principles outlined here apply to other laboratory computerized systems.
Before we can focus on the functions of an ideal CDS, however, we need to set the scene by looking at the role and function of a regulated analytical laboratory and the role of a CDS within it. This is the scope of the first part of this series.
Role of Analysis
The purpose of analysis is to predict the properties of a batch or lot based upon a sample taken from it on a sound scientific basis or a subject concentration time profile from a nonclinical or clinical protocol. Clearly the sample must be representative of the batch, but the issue of sampling and sample management is not considered in this series of articles. Given that business and regulatory decisions are made on the basis of such a prediction, a total data quality management system must be in place to ensure the integrity and security of metrology and derived results at all stages of the analytical process.
There are a number of critical aspects of an ideal quality management system:
• It should follow a life-cycle approach based on the principles of International Conference on Harmonization (ICH) Q10 (3).
• Data acquisition must take place at or close to the point where the data are generated.
• Only electronic interfaces must be permitted; there should be no facility for making manual inputs. Therefore all systems and instruments must be interfaced and integrated together to prevent manual entry or reentry of data followed by subsequent transcription error checking (as occurs with a hybrid system).
• Full and transparent traceability of both the data acquisition and the subsequent calculation processes must be in place.
• The location of the data and the associated metadata and subsequent metadata must be known and secured to enable rapid retrieval.
• Data and metadata must be secure at the time of capture, so that changes can only be made through the application software, with corresponding audit trail entries.
The primary focus of any process control strategy is always the prevention of error from controllable factors. The detection of error from uncontrollable sources is a secondary but important consideration, which we will explore below.
The Analytical Process
The basic analytical procedure is shown in Figure 1. Inputs consist of samples, reagents, standards, and a specific analytical procedure with secured outputs of raw data, metadata, and reportable results. Throughout the execution of any analytical procedure there are controllable and uncontrollable factors to consider.


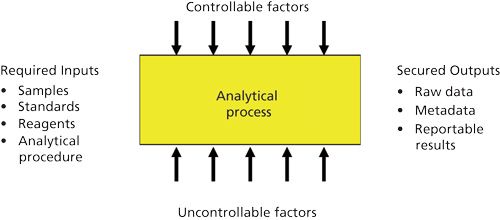
The basic representation illustrated in Figure 1 is simplistic, however. There are two types of life cycles in the use of an analytical procedure: the within-procedure life cycle and the between-procedure life cycle.
The within-procedure life cycle is short term and relates to the performance of the analytical process for an individual analytical measurement sequence. The between-procedure life cycle relates to ongoing verification of a state of control while the procedure is in routine operation and covers monitoring of its performance and changes in terms of time-related shifts and drifts in analytical response. Data from the within-procedure and between-procedure life cycles should be trended (4) to obtain an overview of how an individual procedure is performing over time.
Such a life-cycle concept is consistent with the core needs of ICH Q10 (3) and the Food and Drug Administration (FDA) process validation guidance (5). The key elements of these two documents are as follows:
- Management responsibility
- Understanding and improvement of process performance and product quality
- Continual review and improvement of the pharmaceutical quality system itself
A recent article in Pharmacopeial Forum from a USP Expert Committee has proposed a life-cycle approach to development and verification of an analytical procedure (6) and is shown in Figure 2. This approach starts with an analytical target profile (ATP) that specifies the performance of the required procedure in relation to a target measurement uncertainty amongst other factors.

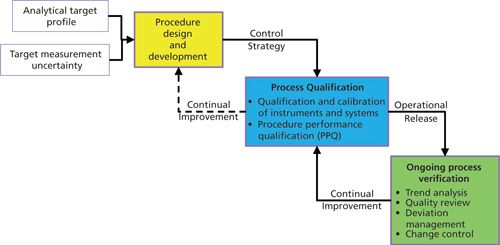
That is to say, the reportable result definition is fit for its intended purpose with respect to a given specification. Crucially, the ATP does not specify the metrological method to be used, only the performance attributes required. An overview of this approach was discussed in a recent “Questions of Quality” column (7). The main change is the inclusion of method development within the scope of analytical procedures where previously it has been excluded, such as in ICH Q2(R1) (8,9).
However the majority of analytical procedures require a degree of specificity (selectivity), and that usually indicates the use of separation techniques such as chromatography. Therefore, if chromatography is to be used for an analytical procedure, any CDS to be used in a regulated laboratory needs to have the functionality to automate this development, qualification, and verification process effectively.
The “Analytical Factory”
The analytical procedure resides within a controlled quality managed laboratory environment: the “analytical factory,” which is shown in Figure 3.

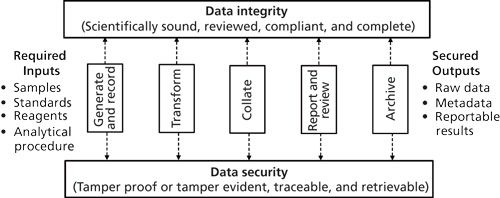
In this figure, the analytical procedure is broken down into the main stages needed to convert the inputs into outputs. Here, the input types are the same as in Figure 1, but we show the process stages familiar to readers-such as “generate and record” (data acquisition), “transform” and “collate” (interpretation), and “report and review”-in more detail. Finally, there is the need for secure and complete delivery of the reportable results. Throughout this process of the analytical factory is the requirement for two main constraints in the process: data integrity and data security. When we mention data we include the associated metadata that put the data in context (this is part of the complete data referred to by FDA regulations [10]). What Figure 3 does not show is the formal destruction of records after the appropriate record retention period has expired.
Metadata are for assessing data integrity. Metadata permit a result to be put into a context; a better term than metadata might be associative or contextual data. The March 2015 Data Integrity Guidance published by the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) has definitions of data, metadata, and primary record. We have written a critique of the definition of primary record, preferring a definition of primary analytical record. (That critique is due for publication in September 2015 [11]). Metadata allow unambiguous definition of the conversion of raw data to reportable values. For example, if presented with a result of 7.0 there is no information about the context of it. Just some of the questions that could be asked are as follows:
- What are the units of measurement?
- What does the result relate to? Does it relate to a batch or experiment number? Or to a stage of manufacturing?
- How was the result obtained? For example, what were the data acquisition method, integration method, instrument control method, sequence file, and any post-run calculations?
- How were the data generated? What instrument, column, and reference standards were used? Who was the analyst?
- What audit trail entries have existed during the analysis, especially around changes and deletions of data? For example, who made the change? What were the original and new values? What was the date and time stamp of the change and reason for the change?
Thus, metadata put an analytical result in context and are critical for ensuring data integrity.
It is important to consider how an analytical procedure relates holistically to the individual components that are controlled and uncontrolled, as suggested by Figure 1. More detail of these two sets of factors is shown in Figure 4, along with their context within an overall quality management system (QMS).

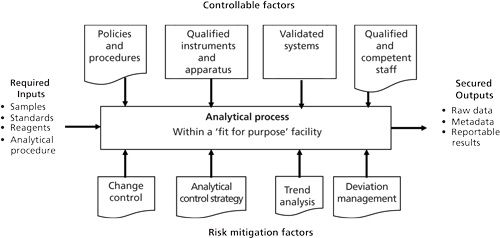
The inputs and outputs shown in Figure 4 are the same constituents as shown in Figures 1 and 3 but the factors affecting their integrity and security are further defined. However, the outputs can also be broken down into further detail covering data and the associated contextual metadata that will be used to generate the output of signed reportable results. Currently, the reportable results can be either the homogeneous electronically signed electronic records or the hybrid of electronic records with signed paper printouts.
The controlled factors are items such as procedures, qualified instruments, validated software systems, validated analytical procedures, and qualified and trained staff. For such factors, a control strategy is required. Uncontrolled factors include deviations from procedures, instrument failures, software and system failures, method drift and uncontrolled changes to analytical procedures, and finally human error. For these factors, an effective detection system is necessary, coupled with appropriate change control procedure and validation where necessary.
An ideal laboratory informatics package of the future should cover the entire holistic analytical life cycle as far as is practically possible without need for additional external systems. Because such a system currently does not exist, we are currently left with an analytical informatics jigsaw puzzle.
Data Integrity Control Strategy
In addition to the analytical procedure life cycles that we have discussed, there also needs to be a control strategy to ensure data integrity within the CDS, as shown in Figure 5. Note that Figure 5 is aligned and consistent with Figure 3.

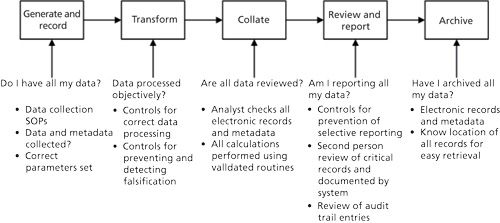
The data integrity control strategy must be integrated into the analytical procedure life cycle. Such integration will provide a mechanism for ensuring that the required data checks are carried out automatically by the CDS as the individual stages of an analytical procedure are executed and will provide evidence to demonstrate that a given procedure is in a state of control.
Summary
Here in the first of this four-part series we have looked at the scope of an ideal chromatography data system or laboratory informatics solution of the future. In addition, there need to be interfaces with other instruments such as analytical balances for direct data acquisition of sample weights into the sequence file to eliminate manual entry and second-person checks of such data.
However, as the focus of this series of papers is the ideal CDS of the future, the remaining parts of this series will only consider an electronic solution: an electronic process that generates electronic records that are signed electronically. The first step in this discussion, in the next article in this series, will consider the architecture of an ideal CDS. Future technical and regulatory-compliant systems will require enhanced functionality to ensure data integrity and security.
Acknowledgments
The authors would like to thank Lorrie Scheussler, Heather Longden, Mark Newton, and Paul Smith for comments and suggestions made during the writing of this series of papers.
References
(1) R.D. McDowall, LCGC Europe27(9), 486-491 (2014).
(2) R.D. McDowall, LCGC Europe28(6) 336-342 (2015).
(3) International Conference on Harmonization, ICH Q10, Pharmaceutical Quality Systems (ICH, Geneva, Switzerland, 2007).
(4) European Commission Health and Consumers Directorate-General, Eudralex: The Rules Governing Medicinal Products in the European Union,Volume 4: Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use; Chapter 6 Quality Control (Brussels, Belgium, 2014).
(5) US Food and Drug Administration, Guidance for Industry: Process Validation (FDA, Rockville, Maryland, 2010).
(6) G.P. Martin et al., Pharmacopeial Forum39(5) (2013).
(7) R.D. McDowall, LCGC Europe27(2), 91-97 (2014).
(8) International Conference on Harmonization, ICH Q2(R1), Validation of Analytical Procedures: Text and Methodology (ICH, Geneva, Switzerland, 2005).
(9) Lifecycle Approach to Validation of Analytical Procedures with Related Statistical Tools, USP Workshop, December 2014 (http://www.usp.org/meetings-courses/workshops/lifecycle-approach-validation-analytical-procedures-related-statistical-tools).
(10) Code of Federal Regulations (CFR), Part 211.194(a), Current Good Manufacturing Practice for Finished Pharmaceutical Products (U.S. Government Printing Office, Washington, DC, 2008).
(11) C. Burgess and R.D. McDowall, LCGC Europe28(9), (2015) in press.
R.D. McDowall is the director of R.D. McDowall Ltd., and Chris Burgess is the managing director of Burgess Analytical Consultancy Ltd. Direct correspondence to:
rdmcdowall@btconnect.com














Characterizing Plant Polysaccharides Using Size-Exclusion Chromatography
April 4th 2025With green chemistry becoming more standardized, Leena Pitkänen of Aalto University analyzed how useful size-exclusion chromatography (SEC) and asymmetric flow field-flow fractionation (AF4) could be in characterizing plant polysaccharides.




Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).

