Hydrophilic-Phase Extraction: A New Avenue of Solid-Phase Extraction for Oligonucleotide Bioanalysis
A novel mode of bioanalytical solid-phase extraction (SPE) has been developed and used for a quantitative application involving an RNA oligonucleotide therapeutic candidate. Previously established protocols and media showed very little recovery, and so this new approach, based on hydrophilic interaction liquid chromatography (HILIC) with an aminopropyl-bonded phase on a silica support and providing an essentially uncharted avenue of selectivity, was explored. The procedure featured only one step prior to sample load, high-organic sorbent conditioning, two washes following the load that switched between low and high pH, followed by elution in an a highly eluotropic HILIC composition. The moniker of “hydrophilic-phase extraction” (HPE) was given to the procedure. The HPE was proven to be a critical part of a fully quantitative HILIC–high-resolution mass spectrometry (HRMS) method for the RNA oligonucleotide and has potential for general application.



Biologics, such as peptides, proteins, and oligonucleotides, are more prevalent as therapeutic candidates than ever. In the exciting arena of oligonucleotide bioanalysis, there are well-known challenges when it comes to any quantitative platform including liquid chromatography–mass spectrometry (LC–MS). The author’s group has recently been focusing on creating quantitative bioanalytical approaches without the use of ion-pairing reagents, and has previously worked with hydrophilic interaction liquid chromatography (HILIC) preceded by polymeric‑based weak cation exchange (WAX) solid-phase extraction (SPE) (1).
This article focuses on the sample preparation and extraction aspect of a recently developed and unique bioanalytical SPE method that also does not require ion-pairing reagents and could possibly be useful for general applicability in analogous applications in the future. Despite the success of the polymeric WAX in the one referenced application (1), and another with a more laborious protocol (2), we found that for one particular application, for an RNA oligonucleotide known as GNV705 AS, we had to find and explore a new realm of selectivity for decent recovery (3). GNV705 AS is the 22-mer antisense strand of a duplex-based RNA therapeutic candidate. Along the GNV705 AS strand, there are numerous methylations and fluorinations, and towards each terminus there are phosphorothioate linkages, typical features for stabilization, and the central region is populated by phosphodiester linkages.
With protocols that were based on references (1) and (2), it was found that recoveries (even after altering the set of key variables in the SPE) were never in excess of 20–30% and, more importantly, were non-reproducible. In the context of oligonucleotide SPE, this ballpark of recovery is not unusually low (4,5), but we wanted as much as possible to achieve concrete method reproducibility and, ultimately, sensitivity. At this point, thoughts turned to producing something distinct on a physicochemical basis but still poised to exploit the innate excellent degree of polarity and ionization of the oligonucleotide analyte. This is even more prominent for RNA compared to DNA because the extra hydroxylation in the ribose moiety offers slightly more polarity.
Considering the pronounced polarity of the silica base, the clear established contribution that it has to HILIC in the LC context (6), and specifically how the highly polar silica chemistry aligns with nucleic acids’ intrinsic deep-rooted polarity, the fundamental affinity for the analyte on such a support should be prominent, in contrast to a polymeric support, which has much more of a hydrophobic constitution. This should be particularly valid if the silica support has a polar bonded phase with an appropriate electrostatic functionality.
In principle, such an idea can certainly work. This group’s own previous research from 2016 demonstrated an application where HILIC has been utilized within the SPE for a bioanalytical application for the therapeutic peptide bivalirudin in human plasma. (7) This produced a high-recovery, validated method.
There was also work reported recently (8) that employed SPE using unmodified silica and HILIC operation, but it was a dual extraction with several liquid–liquid treatment steps in precedence to SPE loading and the remainder of the extraction. This presents an undesirable laborious element and more sources of variability, even though the ultimate extract cleanliness could be excellent. For the work described here, we aspired to a more high-throughput protocol where plasma is simply diluted in the appropriate aqueous diluent prior to loading, more aligned with classical SPE in the regulated pharma industry context.
The key feature was that this SPE would follow a HILIC scheme, with rudimentary polar and electrostatic interactions being harnessed.
Experimental
Chemicals and Materials: The reference material for the analyte GNV705 AS, as well as the N-1 and N-2 metabolite reference materials, were kindly gifted by Genevant Sciences. The structural analogue—incorporating three additional nucleotide units in the middle of the strand—internal standard reference material was obtained from Integrated DNA Technologies. Acetonitrile, concentrated phosphoric acid (85%), concentrated ammonium hydroxide (25%), concentrated formic acid, and ammonium formate were all obtained from Sigma Aldrich and were LC–MS‑grade, with the exception of phosphoric acid, which was ACS-grade. Water was purified in-house with a Thermo Scientific Barnstead Nanopure purification system by reverse-osmosis filtration and subsequent deionization to a resistivity of 18.2 MΩ cm. Control cynomolgus plasma with K2EDTA anticoagulant was obtained from BioIVT, including from six individual donors for differential tests and selectivity.
Calibration Standards and Quality Control Samples: The GNV705 AS primary solution and a candidate internal standard (IS) were prepared at 100 µM in 9:1 (v/v) water–acetonitrile. Calibration quality control (QC) sample spiking solutions and the IS spiking solution were prepared in 9:1 (v/v) water–acetonitrile in polypropylene tubes. Plasma was prepared in analogous polypropylene tubes at concentrations of 0.5, 2, 5, 10, 20, 50, 100, 200, 500, 1000, and 2000 nM for calibrant samples and at 0.5, 2, 5, 10, 20, 100, and 2000 nM for QC samples. Volumes of spiking solution were no more than 2% of the volume of plasma spiked.
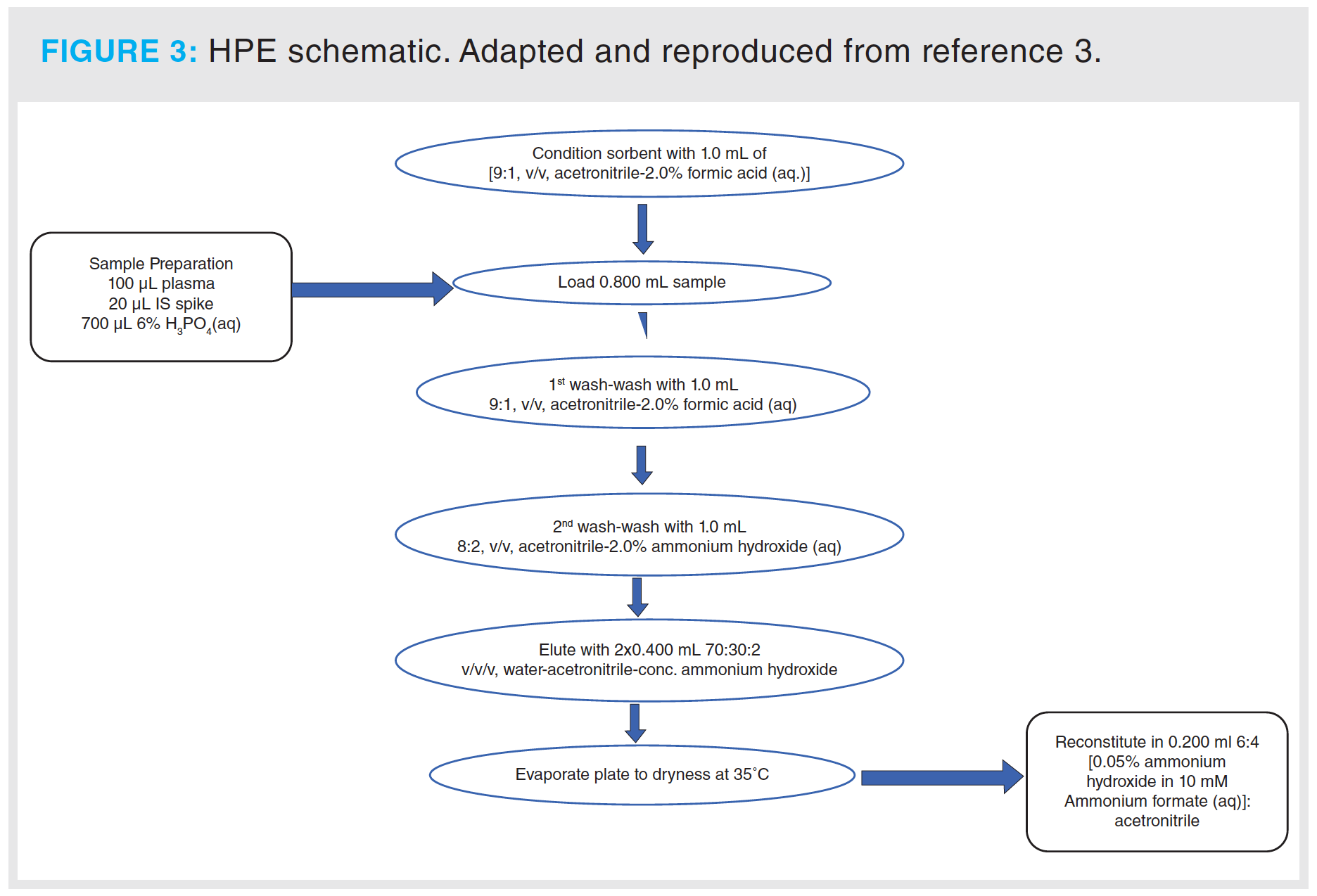
Optimized Sample Preparation: As a final optimized procedure, the solid‑phase extraction of GNV705 AS from cynomolgus plasma was performed as follows. The SPE sorbent was United Chemical Technology NAX, 100 mg, an aminopropyl phase on a silica base, in 96-well format. The 96-well 1-mL round‑bottom collection plates used to receive the final eluent were regular DNA LoBind polypropylene from Eppendorf. Each step where liquid was applied was allowed to pass through gravity alone. The time taken was three to four minutes for this to occur, corresponding to an adequately slow linear flow velocity conducive to best performance under the circumstances of high-energy ionic interactions being formed and broken.
The candidate IS, in 1:9 (v/v) acetonitrile–water at 2500 nM, was added in 20 μL aliquots to 100 μL plasma within 1.5 mL regular polypropylene tubes. This resulted in an internal standard concentration of 500 nM in matrix. Then, a 2-s vortex of each tube took place. This was followed by the addition of 700 μL 6% H3PO4 (aq.) to each sample and another vortex step.
Sorbent conditioning involved the application of 1 mL 9:1 (v/v) acetonitrile–(2% formic acid [aq.]), and there was no subsequent equilibration step prior to loading the diluted plasma sample. The prepared samples (800 μL) were loaded onto the conditioned sorbent beds. This was followed by the application of a 1 mL wash with 9:1 (v/v) acetonitrile–(2% formic acid [aq.]). The next wash was with 1 mL 8:2 (v/v) acetonitrile–(2% ammonium hydroxide [aq.]). Analyte elution was affected by the application of 2 × 400 μL 2% ammonium hydroxide in (3:7 acetonitrile–water) into the 1-mL round-well 96-well collection plate. Finally, the eluates were evaporated under oxygen-free nitrogen at 40 °C and then reconstituted in 200 μL 4:6 (v/v) acetonitrile–(0.05% ammonium hydroxide in 10 mM ammonium formate [aq.]). The block was then sealed, put on a plate shaker at 500 rpm for 10 min, before being placed in the autosampler compartment at 10 °C awaiting injection.
Liquid Chromatography: The analytical column for the GNV705 AS quantitative method was a 2.1 × 50 mm, 1.7-μm Waters Acquity Premier UPLC BEH Amide. The LC front-end system was a classic Waters Acquity UPLC unit that included pump, degasser, autosampler, column heater, and mobile phase pre‑heater. The autosampler compartment was maintained at 10 °C. Gradient elution was employed, with mobile phase components of 0.05% ammonium hydroxide in 10 mM ammonium formate (aq.), pH measured at 9.1, and acetonitrile, delivered at 0.45 mL/min and with mobile and stationary phases at 40 °C. For each gradient cycle, initially the mobile phase composition began at 20% aqueous, and the mobile phase composition underwent a linear excursion over the next 5.0 min to 50% aqueous. This composition was held for 0.5 min and then re-equilibration took place over the remaining 1.0 min of the 6.5 min overall runtime. The injection volume was 6.0 μL and partial loop with needle overfill (PLNO) mode was used, along with a strong wash composition of 0.05% ammonium hydroxide in 10 mM ammonium formate (aq.) and a weak wash composition of 1:1 (v/v) acetonitrile–(0.05% ammonium hydroxide in 10 mM ammonium formate [aq.]).
Mass Spectral Detection: The high resolution mass spectrometer was a Sciex ZenoTOF 7600 System equipped with an Optiflow ion source and auxiliary gas heated to 550 °C, with no split of the LC flow into the source. A negative MRMHR acquisition was applied for quantification, where the precursor ion m/z target was set as 1832.3 and a
MS/MS scan range was 100–2000 m/z. The extracted ion chromatograms (XICs) of transitions 1832 " 586.1134, 1832 " 664.0810, and 1832 " 604.1199, where all values denote m/z, were generated using Sciex OS software with the extraction window width set at 0.02 Da. The precursor ion involved was the same as for nominal mass, corresponding to the charge state of [M-4H]4-. The selected product ions were analogous to the nominal mass scheme. Again, these peaks were selected on the basis of most compelling intensity and verifiability. The peak area-based integration for the quantitative endpoint involved the summation of the peaks in each transition.
Results and Discussion
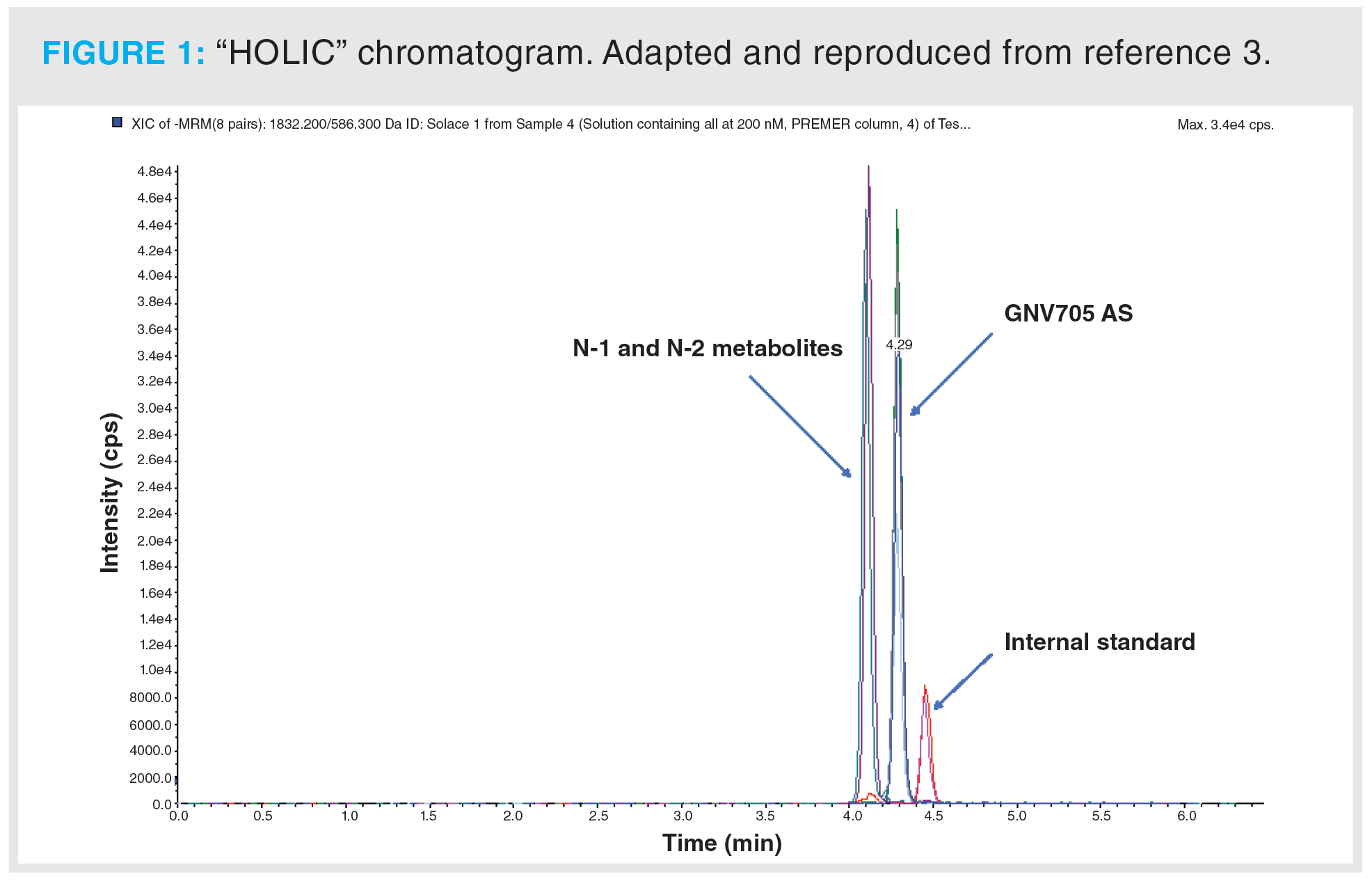
HOLIC: To dwell briefly on the chromatography, the HILIC nature of the dynamics was a critical component of the methodology in terms of the requirement for no ion-pairing reagents to be present. The setup just described represents the bones of what we have assigned the moniker “HOLIC” (3), a play on the HILIC abbreviation interfaced with the term “oligo”. We felt it was merited particularly on the basis of observed applicability to a wide range of different oligonucleotide methods as performed in our laboratory (data not shown). A representative chromatogram is shown in Figure 1. The baseline resolution of the analyte GNV705 AS from the earlier-eluting N-1 and N-2 metabolite pair is evident, and the similar resolution from the internal standard, which elutes last.

Hydrophilic-Phase Extraction: The extraction is clearly the centrepiece of this article. In the previous SPE work of fundamentally similar nature reported for the therapeutic peptide bivalirudin (7), HILIC was proven to be the critical functional mode throughout the bulk of the SPE process, and for this new oligonucleotide application, the anticipation and outcome were no different. This time, instead of the cation exchanger used for the peptide capture and subsequent release, an aminopropyl function was used, appropriate for the negative charge strongly and abundantly present with oligonucleotides. All things considered, at the pivotal load step it was ideal for the most efficient capture. In the absence of the classical partitioning HILIC model because of the high aqueous content—in terms of the fundamental physicochemical affinity as alluded to earlier—the profound polarity of the silica support in conjunction with the short-chain charged bonded phase matched the acute polarity of the RNA analyte.
The initial attempts using a 100 mg bed weight of the aminopropyl phase on a silica base brought improved recoveries from what we had seen at any point prior in this project. From then, it was a straightforward series of experiments to fully optimize the procedure.
For the 100 mg packed sorbent, a procedure prior to elution, and the sample load notwithstanding, based on 1 mL solvent application at each step was appropriate. In all steps, a minimum 10% aqueous was maintained to preserve HILIC conditions and avoid oligonucleotide precipitation via lack of organic solubility that may lead to loss of signal and imprecision. In a unique and interesting discovery, it was found that omitting the high-aqueous equilibration step—as would be intuitive in almost all SPE procedures—was found to improve recovery in this process. This was postulated to be an artefact of the retained oligonucleotide band being so hydrophilic as to be repelled from the surrounding organic environment that would be present after the conditioning step, thereby not broadening as it otherwise would after a high-aqueous step and avoiding associated recovery loss. Similarly, an initial fully aqueous wash step was omitted to avoid breakthrough in the highly aqueous conditions that fully solubilize nucleic acids such as the analyte.
The single condition was therefore 1 mL of 9:1 (v/v) acetonitrile–(2% formic acid [aq.]), with the acidity maintaining the ionized state of the key ionic moiety of the bonded phase. The wash after the sample load was identical to the condition—again the high “oligo-phobic” organic helping to prevent analyte breakthrough.
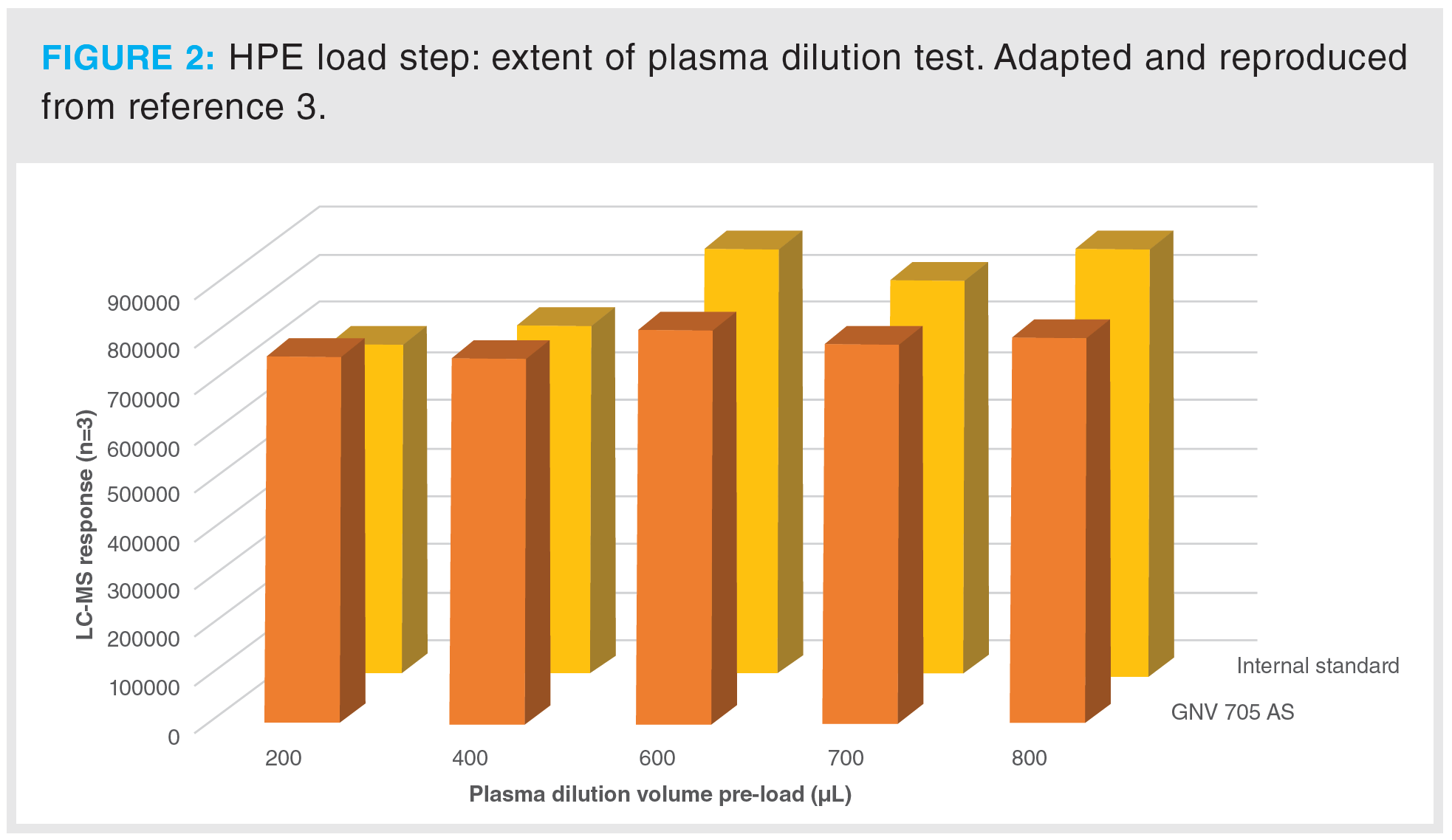
For the plasma sample dilution and loading, initially it was decided to use 6% phosphoric acid (aq.) as the diluent, a little more concentrated than what we had used in previous work (1) to better align with the low plateau of pH attained by phosphoric acid with increasing concentration. This did appear to make a difference for the first few moments where the high-energy electrostatic bonds are forming, albeit involving a little more neutralization of the phosphate backbone and accordingly more nonpolar interactions, low energy thus rapidly manifesting, a phenomenon synonymous with success at this stage of classical mixed-mode SPE. A brief test (data not shown) showed around 25% better recovery in going from 4% to 6% phosphoric acid in the otherwise established conditions. On this note, further work looking into incrementally higher concentrations, up to perhaps 10% at this stage, is worthwhile. Figure 2 shows the data for both analyte and internal standard from a test where the 100 μL plasma volume was diluted to different extents prior to loading; in at least one separate oligonucleotide client-related application a dependency in recovery was established (data not shown) in the sense of recovery increasing with total volume (plasma and diluent) applied until a maximum was reached. The reasoning is speculated to be to do with maximizing opportunity for capture by spacing out volumetrically the species to be retained under a flow that is as slow as is practically reasonable because, as mentioned, high-energy electrostatic bonding needs to manifest. Gravity alone gave suitable flow for this work, with the packing of the wells allowing flow at each step, which came to 4 min at the most. With regards to the outcome of the test, as shown in Figure 2, any such dependency did not seem overwhelming for GNV705 AS, but was more apparent for the IS, showing a quite definitive signal increase backed up by triplicate analysis. We settled on the highest value of 800 μL for safety and practicality.

As mentioned, the first wash was identical to the single-step condition, 1 mL of 9:1 (v/v) acetonitrile–(2% formic acid [aq.]). The following wash immediately prior to elution was with 1 mL 8:2 (v/v) acetonitrile–[2% ammonium hydroxide (aq.)]. This introduced a little eluotropicity by increasing in aqueous content and featuring a slightly alkaline pH; without enough alkalinity to neutralize the key ionic moiety in the bonded phase, particularly in the high-acetonitrile conditions, retention persisted. Elution was applied, with 2% ammonium hydroxide in [3:7 acetonitrile–water], highly aqueous to fully solubilize the analyte, and sufficiently alkaline, with a pH of 11.5 as measured in the aqueous component, to neutralize the sorbent. For the 100 mg packed bed, we anticipated a void volume of 150 μL and thus we included a test with 200 μL total volume. This was not enough to effect full elution. Only once 2–3 void volumes had passed—a total of 400 μL—did we see the full response corresponding to the elution of what remains on-cartridge. To err on the side of caution, and with knowledge of how incurred samples can differ quite profoundly in their natural matrix compositions, we decided to establish the final elution volume as 800 μL total to afford sufficient eluting power in unusual conditions that may demand it.
Decisively, in all the available numerical detail over the low QC (LQC) and high QC (HQC) nominal levels, the recovery for GNV705 AS was above 60% and the candidate internal standard was in the same region as calculated over the replicate set at the associated one nominal level. Specifically, the analyte recovery was 64.1% and 62.0% at the LQC and HQC levels, respectively, and 57.4% for the internal standard.
It may be speculated that a marriage between a HILIC-based sample extraction and a HILIC LC–MS analytical endpoint may not result in clear orthogonality of selectivity between these two methodological components. However, we believe that this ever-desirable feature is actually largely preserved because of the details in each case. Primarily, the chemistry of the stationary support was different between the analytical column and the solid-phase packed cartridges—amide on hybrid polymer-silica compared to aminopropyl on silica. The extraction used a comprehensive wash scheme involving both acidic and alkaline applications prior to elution, which is almost unique apart from the portfolio of our own work (1,3,7), and will have a large bearing on selectivity and the elimination or resolution of interferences. The performance data are impressive, underlining the adequate selectivity.
The final HPE schematic is shown in Figure 3. It is not difficult to see that we have a liking for monikers in this group, and for this we have used “hydrophilic phase extraction” (HPE), since it can be none other than hydrophilic interactions manifesting at the key stages of the process, for the reasons outlined.

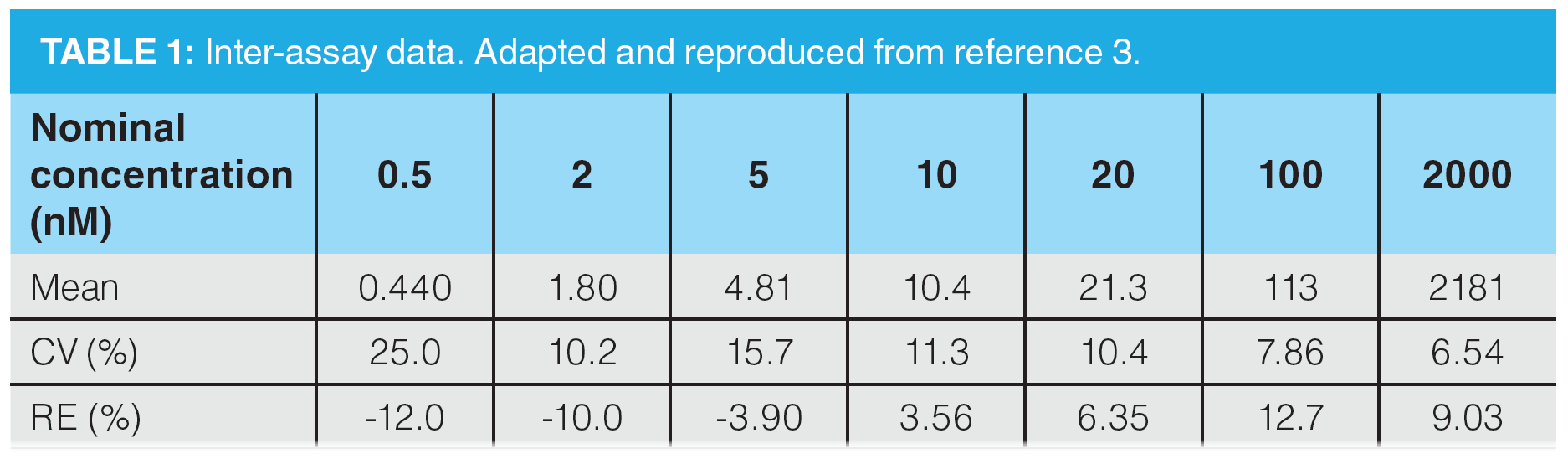
Inter-Day Analysis: To demonstrate quantitative method ruggedness, three inter-day analytical batches were performed. From these, the bias and precision ranged from -12.0% to 12.7%, and 6.54% to 25.0%, respectively, over the calibration range of 500 pM to 2000 nM. The inter-day performance data are shown in Table 1.

Outlook: Over the decades, silica‑based SPE has become less popular in regulated bioanalysis, while the popularity of polymeric products has risen accordingly. However, particularly with the importance of SPE for reliable quantification of biologics, this work may help reopen the door for the silica base in such applications, with sound underlying chemistry-based reasons. Silica may find a niche to be used—in conjunction with polar and ionizable bonded phases—in HILIC mode, distanced from reversed-phase operation, which has dominated the history of silica in bioanalytical application. This could apply to all biologic-based analytes with an LC–MS bioanalytical endpoint, in light of the accompanying polarity at hand.
Conclusion
Within the development of a quantitative bioanalytical LC–MS method for an antisense RNA oligonucleotide, and within the valuable theme of maintaining a complete absence of ion-pairing reagents, a novel mode of SPE has been developed and used for this, and the basic framework likely has a great deal of general utility. It is distinct from regular means of bioanalytical SPE in that HILIC is principally manifest and governs the band migration along with ion-exchange via the aminopropyl functionality. Only one condition step was used and, after sample load, a double-wash regime was applied at both low and high pH, thereby stepping up the aqueous and the basicity to finally effect elution. We have given the extraction the moniker “HPE”, hydrophilic-phase extraction. The method was proven to be rugged and reliable over a sequence of analyses using an analytical endpoint featuring our default HILIC setup, “HOLIC”, interfaced with the latest in accurate mass detection.
References
- R. MacNeill, T. Hutchinson, V. Acharya, R. Stromeyer, and S. Ohorodnik, Bioanalysis 11(12), 1155–1167 (2019).
- “Oligonucleotide purification products: OTX phase detail”: https://www.phenomenex.com/Products/Detail/Clarity%20(SPE)/OTX
- P. Anand, M. Koleto, D. Kandula, L. Xiong, and R. MacNeill, Bioanalysis 14(1), 47–62 (2022).
- W. van Dongen and W. Niessen, Bioanalysis 3(5), 541–564 (2011).
- L. Nuckowski, A. Kaczmarkiewicz, and S. Studzińka, J. Chromatogr. B 1090, 90–100 (2018).
- A. Alpert, J. Chromatogr. 499, 177 (1990).
- R. MacNeill, R.V. Acharya, N. Patel, G. Parisi, K. Katto, and R. Lovich, Bioanalysis 8(21), 2195–2203 (2016).
- L. Nuckowski, L.A. Kilanowska, and S. Studzińska, J. Chrom. Sci. 4(58), 383–387 (2020).
Robert MacNeill is Head of Method Development at LabCorp Drug Development. He received his bachelor’s degree with honours in chemistry from Heriot-Watt University, and his M.Sc. in analytical chemistry from the University of Huddersfield, UK. Robert is also a Chartered Chemist and Fellow of the Royal Society of Chemistry, and has 24 years of experience in all aspects of quantitative bioanalytical LCÐMS method developmentÑwith 15 of these spent heading method development activities within LabCorp Drug Development at the Somerset (New Jersey, USA) site. Taking a prominent role in innovation projects with related R&D activities, Robert has been an active contributor to the scientific literature for many years.




 for Use in LC")


In the present study, a gradient reversed-phase high-performance liquid chromatography (RP-HPLC) method has been designed and validated to quantify ornidazole (OZ) in the marketed formulation (oral gel) with the application of QbD.


A column with chemically modified column hardware showed improvements in analytical performance for siRNA compared to a conventional stainless-steel column.




