HPLC–SPE–NMR — A Novel Hyphenation Technique
LCGC Europe
High performance liquid chromatography–solid phase extraction–nuclear magnetic resonance (HPLC–SPE–NMR) is a novel hyphenation technology that concentrates single chromatographic peaks to elution volumes matching those of NMR flow probes. The SPE unit facilitates the solvent exchange from the mobile phase of the optimized HPLC assay to a deuterated NMR solvent. The well-defined NMR solvent conditions make spectra comparisons feasible, which means databases and spectra catalogues can be used to swiftly identify analytes. The ability to accumulate analytes on the SPE cartridges by multiple trapping reduces the need to perform residual solvent suppression experiments and allows heteronuclear NMR experiments to be performed overnight. Structure elucidation of natural products directly from crude extract HPLC samples has become the key application of this technique.
Christoph Seger and Sonja Sturm, Institute of Pharmacy/Pharmacognosy, Centre of Molecular Biosciences, University of Innsbruck, Austria.a
Nuclear magnetic resonance (NMR) spectroscopy is an indispensable analytical tool to characterize the structure of organic molecules ranging from small and simple analytes to high-molecular-weight biopolymers, such as peptides, proteins and oligo-nucleotides (RNA and DNA). Its major advantage over mass spectrometry (MS)-based techniques is the potential to analyse molecular scaffolds on the level of atom–atom connectivities (bonds) by establishing unequivocal homonuclear (usually 1 H–1 H) or heteronuclear (1 H–13 C, 1 H–15 N, 1 H–31 P, 13 C–31 P) neighbourhood relationships in a non-destructive way.



NMR is outstanding in terms of structural information gained but is less sensitive than other spectroscopy and spectrometry methods, including mass spectrometry (MS). NMR instruments and sample preparation techniques are constantly being pushed to their limits to narrow this gap. High-resolution magnets, which increase the signal strengths, and can reach 22.3 T (equalling a 950 MHZ 1 H resonance frequency) alongside the introduction of liquid helium-cooled NMR probes (decreasing the thermal noise) have been the main focus of recent instrument research.
Sophisticated strategies to replace the natural isotope distribution with only a small fraction of NMR-detectable atoms in a molecule (1.1 % 13C, 0.3 % 15N) by up to 100% of the desired stable isotope have become indispensable sample preparation protocols for biomacromolecular NMR investigations. However, when working with small molecules, particularly if NMR is used to elucidate the structure of natural products or compounds formed during industrial organic synthesis, this approach is usually not feasible due to economical limitations of using pure stable isotopes materials throughout (bio)synthesis.


Keynotes
HPLC–NMR Hyphenations
The pioneering contributions of Klaus Albrecht1,2 which combined LC-based analyte separations with NMR detectors (LC–NMR), were a great leap forward in the analysis of complex samples of limited quantitative availability (mass-limited samples).3–5 Two major hyphenation strategies emerged with HPLC–NMR probes either matching analytical HPLC dimensions or being adjusted to capillary HPLC flow regimes.
In HPLC–NMR probe designs with conventional saddle coils and flow cell volumes — currently ranging from 60–250 μL — are used. Experimental setups gradually evolved from on-flow experiments to stopped-flow LC–NMR assisted by the temporal segregation of chromatography and NMR using loop collection and loop storage devices for peak parking. Commercial HPLC–NMR setups are available from all major NMR machine producers. The application of HPLC–SPE–NMR emerged from this idea. In a pioneering publication published ten years ago Griffiths and Horton described a post-HPLC column trapping setup using a guard column as the analyte-enrichment device.6
This hyphenation set-up allowed
- on-line trapping of the analyte peak of interest eluting from a HPLC system on a pre-equilibrated guard column cartridge
- HPLC mobile phase to be removed by prolonged washing with a solvent of low elutropic strengths (e.g., D2O or H2O)
- analytes to be backflushed from the solid phase to the NMR flow probe with a deuterated NMR solvent of sufficient elution power ( e.g., CD3OD (deuterated methanol), CD3CN (deuterated acetonitrile)).
They also proved that the observed signal to noise ratio (S/N) gains were inversely proportional to the chromatographic peak (peak volume). Consequently they reasoned, that reducing the elution volume of an analyte close to the flow cell volume of the LC–NMR probe head resulted in optimal sensitivity enhancement. Their conclusions can still be considered major keystones in the development of HPLC–SPE–NMR.
Generally, analyte release from the SPE device has to be optimized so that a narrow elution band is formed and further peak diffusion on the way to the detection cell is minimized. In two earlier contributions dealing with the analysis of secondary plant metabolites, Nyberg et al. used a SPE–NMR setup to transfer analytes containing semi-preparative HPLC fractions to a NMR spectrometer. In their first contribution, the authors obtained complete sets of 2D homo-and heteronuclear NMR spectra of < 0.5 mg substance (capsaicinoides) in pure CD3OD within a few hours, proving that this hyphenation setup had a strong potential for the de novo structure characterization of natural products.7
In the second contribution a series of structurally demanding saponine congeners have been addressed. Their structural characterization was aided by matrix assisted laser desorption time-of-flight mass spectrometry (MALDI–TOF MS) restricting the number of possible isomers to a few for each analyte. Structure reporter groups, mainly 1 H-NMR signals of the anomeric CH groups, were used in a comparative manner to characterize the carbohydrate side chains.8 The authors proved that the need of direct, unequivocal, and unquestioned comparability of NMR spectra for the structural characterization of complex natural products can be met by SPE–NMR by the complete solvent exchange from the HPLC mobile phase mixture to a well-defined NMR solvent.
About six years ago an automated and refined HPLC–SPE–NMR setup allowing full control over (a) HPLC–UV or HPLC–MS triggered trapping events; (b) SPE cartridge handling in a 96 well-plate format; and (c) analyte elution from the SPE to a NMR spectrometer or another collecting device, such as an autosampler or NMR tubes, was introduced by one of the major NMR manufactures. Currently, this HPLC–SPE–NMR platform (Figure 1) is used by most of the laboratories, although other configurations have been reported, too.9–11

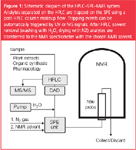
Figure 1
If NMR spectrometers from different producers or of different field strengths are available in a laboratory, HPLC–SPE–NMR tends to be used as offline HPLC–SPE tube-NMR combination.12–14 This means that the advantage of transferring a chromatographic peak to a NMR solvent in an automated, reproducible, and conserving manner is combined with the flexibility of tube NMR, which allows the use of the optimal spectrometer available. Numerous hardware changes for the commercialized platform, resulting in improved performance of the critical SPE step, have been reported by a Norwegian team.15
Following the evolution of capillary HPLC for the analysis of mass-limited samples, capillary NMR probes with solenoidal radio frequency (RF) coils have been developed.16,17 This approach resulted in the design and commercial introduction of probes with an active sample volume (capillary volume within the RF sender/receiver coil) of 1.5 μL.
Although capillary NMR can be successfully hyphenated to the matching HPLC equipment,18,19 or combined with sample robots in industrial-style high-throughput setups,5 it is mostly used in an off-line mode. Dried samples are taken up in few microlitres of NMR solvent and introduced into the probe manually or by the aid of a syringe pump. This setup has been successfully used to characterize mass limited natural product samples20,21 within decent NMR acquisition times (e.g., acquisition of COSY, HSQC and HMBC spectra of ;100 nmol overnight).22 This brings these probe heads in the same sensitivity range as state-of-the-art HPLC–SPE–NMR equipment.
Advantages and Limitations
As mentioned previously, post-HPLC focusing of analyte peaks to match the volume of the NMR probe flow cell by the aid of the SPE-based trapping device is the major advantage of HPLC–SPE–NMR. However, this makes optimal SPE trapping and elution conditions necessary which have to be optimized at least for each analyte class on a instance-to-instance basis. In the trapping process, the amount and composition of the post-HPLC added makeup flow used to promote the analyte binding onto the SPE stationary phase is a promising candidate parameter for assay improvements. Analyte release from the SPE is strongly influenced by both the elutropic power and the hydrogen bonding capacity of the NMR solvent, making both acetonitrile and methanol prime candidates.
Because basic rules of chromatography (e.g., mobile/stationary phase dependency of analyte retention factors, plate number of sorbent materials, break through characteristics of SPE stationary phases) do apply, knowledge from conventional and well-established off-line SPE approaches23 can be helpful to find decent working conditions.
Divenylbenzene (DVB)-type polymers and RP–C18 silica stationary phases are commonly used as stationary SPE phases with 1–2 mL/min H2O as post-HPLC make-up solvent. These conditions have worked for most published applications. However, limitations can be expected for charged or polar analytes such as alkaloids or organic acids. Modified SPE phases (i.e., SAX or SCX materials or porous carbon materials) could be useful in these applications.9,15,24,25 Multiple trapping of the analytes onto a single SPE cartridge is feasible (Figure 2), however, pronounced differences in the trapping efficacy between the available stationary phase materials have been observed.26–28 In one study, ; 100 μg scopoletin have been accumulated on a 2 × 10 mm GP phase cartridge (Spark, Emmen, Holland) by seven trapping steps, whereas only ; 20 μg was retained on a C–18 column under identical trapping conditions.27

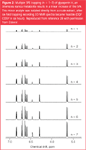
Figure 2
Generally, with the multiple trapping strategy several dozen micrograms of an analyte can be transferred from the SPE to the NMR spectrometer. This allows recording of two-dimensional (2D) NMR experiments mandatory for successful de-novo structure elucidation within a few hours on a standard NMR spectrometer (Figure 3). In the majority of applications, CD3CN (deuterated acetonitrile) and CD3OD (deuterated methanol) were used to elute the analytes from the SPE cartridge. Deuterated chloroform (CDCl3) — a widely-used NMR solvent — has rarely been used. Highly viscose NMR solvents, such as DMSO-d6 (deuterated DMSO) or C5D5N (deuterated pyridine), which are commonly applied in sample tube-based NMR work, have not yet found their way to HPLC–SPE–NMR. Less than 1 mL NMR solvent per analyte is needed, including all equilibration and washing steps. Consequently, a significant reduction in consumable costs can be achieved compared to LC–NMR, where the use of deuterated organic solvents is impossible for reasons of expense.


Figure 3
Furthermore, deuterated NMR solvents reduce the need of solvent signal suppression, which usually leads to the loss of valuable spectral information in the vicinity of the solvent signals in LC–NMR. If multiple SPE trapping is employed, 1 H NMR spectra can be even recorded without solvent suppression. Consequently, a significant improvement in the quality of the NMR spectra obtained is observed and is comparable with sample-tube-recorded NMR spectra. As mentioned previously, the solvent exchange in HPLC–SPE–NMR ensures the comparability of the resulting NMR spectra, even if varying HPLC mobile phase conditions were used to separate the analytes before the trapping step.
This is the basis of direct spectra comparison, which is essential in NMR-based structure analysis, especially if unknown analytes have to be characterized from scratch — a situation frequently encountered in natural product analysis. In addition, separating the NMR with its special solvent requirements from the HPLC by the SPE device allows HPLC separations to be performed under optimized conditions.
Mobile phase systems routinely used for high performance liquid chromatography diode array detection mass spectrometry mass spectrometry (HPLC–DAD–MS/MS) setups can be used for HPLC–SPE–NMR. The use of any buffer additive or solvent mixture is possible, although volatile additives showing no — or hardly any — NMR signals (e.g. formic acid, acetic acid and their volatile ammonium salts) should be preferred to avoid precipitations in valves and capillaries. As a result of the possibility of multiple SPE trappings the application of semi-preparative HPLC equipment is generally not necessary.25
The sensitivity of the HPLC–SPE–NMR is comparable with capillary NMR in routine natural product analysis setups.22 To investigate a typical secondary metabolite (molecular weight < 700 Da), about 2 μg analyte/μL NMR solvent have to be transferred to the active HPLC–SPE–NMR probe volume (i.e., 120 μg analyte in a 60 μL flow probe with a 30 μL active cell volume) of a 600 MHz NMR spectrometer equipped with a room temperature probe head to obtain a series of high-quality homonuclear and heteronuclear 2D spectra (e.g., a combination of DQF-COSY (Double-Quantum-Filtered Correlation Spectroscopy), TOCSY (Total Correlation Spectroscopy), HSQC (Heteronuclear Single Quantum Correlation), and HMBC (Heteronuclear Multiple-Bond Correlation) overnight.
If only 1 H NMR spectra need to be recorded, sufficiently good spectrum qualities (i.e., visibility of all signals including CH2 multiplets to allow the interpretation of H–H coupling) can be obtained from about 10 nmol substance (i.e., 5 μg at 500 Da) within a reasonable timeframe (Figure 4).7,29 Nevertheless, capillary NMR probes show better mass sensitivities because of their special design.16,17


Figure 4
Applications
Currently about three dozen HPLC–SPE–NMR applications have been published (Table 1). Not surprisingly, most of the work was devoted to the characterization of secondary natural products from different sources. Analyte encompass a broad range of polarities and structural diversity; they include polyphenols (i.e. flavonoids, isoflavonoids, phenolic acids),12,25,28,30–35 coumarins,27 iridoids,29,34 terpenoids, 3,25,33,34,37–39 cardenolides, saponins,8 polyacetylenes,41 lignans,42 and some alkaloids.24,43,44 The investigated plants ranged from widely applied phytopharmaceuticals as Harpagophytum procumbens (Devils claw)29,33,34 or Hypericum perforatum (St. John's Wort)32,35 and plant products like olive oil,31 to less investigated systems like Smirnovia iranica,28 Kahania laniflora,40 Corydalis solida,44 or even polymeric dissolved natural organic matter (DNOM).45

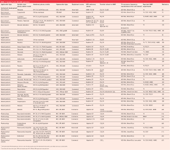
Table 1: Overview of HPLCâSPEâNMR applications.
Two major approaches regarding sample preparation were observed.
Crude extracts were utilized if comprehensive series of major metabolites had to be characterized from a single sample,12,25,28,30,32–34,38,40 whereas sample enrichment strategies — which can include a range of column chromatography techniques, preparative HPLC or offline SPE — were used to focus on minor constituents.7,8,24,29,36,41,44
Both approaches usually use analytical chromatography assays prior to analyte transfer to the SPE. Generally about 1 mg of the crude sample mixture was injected onto the HPLC column. Different mobile and stationary phases were used with a special emphasis on RP–C18 columns and acidified water/acetonitrile gradients. Presented HPLC assay times usually exceed one hour if crude extracts were addressed; the sub-fraction approach generally resulted in shorter HPLC assays. Single or multiple analyte peak trapping was used. In the later instance 2D homo-and heteronuclear NMR data were frequently recorded to support the structure elucidation process.27,28,30,33,34,38,40
The investigated major and minor constituents have been unequivocally characterized by the gathered NMR and MS/MS data and even novel derivatives have been successfully described. In the instance of H. procumbens, 23 secondary metabolites have been identified by HPLC–SPE–NMR.29,33,34 Working with crude extracts several major known phenylethanoid and iridoid congeners and a series of diterpenes, including a structurally complex novel Diels–Alder dimer, have been identified by applying multiple analyte trapping and recording a series of 2D NMR spectra.33,34 Using a more targeted approach to investigate a sub-fraction of a chromatographic workup of a crude extract, four isobaric iridoid regioisomers were characterized. As a result of the preparative analyte enrichment prior to HPLC–SPE–NMR experiments, single SPE trapping was sufficient to obtain 1 H NMR spectra. Unequivocal analyte identification was possible and a novel iridoid congener was identified.29
Ten out of seventeen characterized isoflavonoids were found to be novel natural products in Smirnovia iranica,28 a hardly investigated species.
In a recent contribution from the same research group, HPLC–SPE as analyte enrichment step was combined with capillary NMR for spectral analysis.38 Since washed and dried SPE cartridges can serve as excellent storage media for HPLC-purified analytes,8 sample transport between the laboratories participating in this study was possible. Analyte elution to the capillary NMR probe with 12 μL deuterated methanol was assisted by the use of a SPE cartridge holder. Possible sources of analyte contamination and breakdown as a result of drying HPLC fractions and manually redissolving the residue in minute ammounts (< 10 μL) of NMR solvent prior to the injection into the capillary NMR probes can be avoided with this setup.
Nine major and minor analytes (phenylpropanoids and sesquiterpene-lactones) of Thapsia garganica have been characterized, proving the compatibility of both technologies.38 In pharmacological-orientated applications trapping of drugs and drug metabolites from biofluids has been reported14,46 as well as the identification of impurities and breakdown products of pharmaceuticals.10,26,47 The structural characterization of reaction products from chemical synthesis can be regarded another feasible application field of HPLC–SPE–NMR (Figure 4).26,41 Several contributions have been devoted to the design of alternative SPE trapping units.9–11
Conclusions
Among the many ways of combining chromatography as sample preparation and analyte separation step to NMR spectroscopy, essential to characterize the structure of organic analytes, HPLC–SPE–NMR is an outstanding technology. It is capable of concentrating chromatographic peaks to elution volumes matching NMR flow probes. This means that even chromatographic separation systems with non-ideal peak shapes (e,g., as a result of sample overload or to other limitations of the assay as often observed for alkaloids) can be successfully transferred to the NMR machine. The possibility of accumulating analytes on the SPE cartridges by multiple trapping allows heteronuclear NMR shift correlation experiments to be obtained overnight (e.g., HSQC, HMBC).
In contrast to capillary NMR, which is often named as an alternative to HPLC–SPE–NMR, the analytes under investigation never leave the closed environment of the HPLC–SPE–NMR instrument. Any analyte manipulations can be controlled, sample contaminations can be easily avoided. Impurities from previous preparative chromatographic operations (e.g., salts, acids) are usually removed by the trapping step. This is certainly an advantage compared with campillary NMR approaches, which rely on analytes purified by offline methods and concentrated several-fold during the redissolvation step to NMR solvent volumes < 10 μL.
Natural product analyses are currently the major application of HPLC–SPE–NMR. Several dozen analytes have been successfully characterized by this setup. The gain in overall analysis speed is dramatic. Particularly, the confirmative HPLC–SPE–NMR analysis of known abundant secondary metabolites in uninvestigated plant species saves personnel and consumable costs because the preparation of milligramme quantities needed for conventional tube NMR can be avoided. As a result of well-defined NMR solvent conditions in HPLC–SPE–NMR, spectra comparisons are feasible; databases and spectra catalogues can be used for swift analyte identification.
For industrial setups, the HPLC–SPE–NMR hyphenation is often reduced to a HPLC–SPE hyphenation to decouple HPLC-based analyte purification and NMR spectroscopy. Enriched SPE-trapped chromatographic peaks are transferred to the NMR solvent and, subsequently, eluted to low-volume NMR tubes matching the SPE elution volume. This allows to transfer the sample to any NMR machine available, for example one being equipped with a cryongenically cooled probe head or one operating at a higher field strength.
HPLC–SPE–NMR covers a broad field of applications. Its impact on accelerating the dereplication process of complex secondary metabolite mixtures is unquestioned. Combining HPLC–SPE–NMR with simple sample enrichment techniques will also be useful for pharmaceutical impurity analysis..
We believe HPLC–SPE–NMR will be soon widely recognized as a novel key technology for unequivocal analyte identification — the prerequisite for successful qualitative and quantitative HPLC–DAD–MS/MS assay development in industry and academia.
Sonja Sturm is assistant professor in the Institute of Pharmacy/Pharmacognosy at the Center of Molecular Biosciences (CMBI) of the University of Innsbruck, Austria. Her research interest focuses on the development and validation of analytical methods for the analysis of pharmacologically relevant natural products in phyto-pharmaceuticals and medicinal plants utilizing modern hyphenated platforms like CE–MS–MS, HPLC–MS–MS and most recently LC–SPE–NMR.
Christoph Seger is independent researcher at the Institute of Pharmacy/Pharmacognosy at the Center of Molecular Biosciences (CMBI) of the University of Innsbruck, Austria. His research focus encompasses NMR based structure elucidation of natural products, LC–SPE–NMR as well as assay development/validation of hyphenated chromatographic methods for routine analysis.
References
1. K. Albert, J. Chromatogr. A, 856(1–2), 199–211 (1999).
2. K. Albert, On-Line LC-NMR and Related Techniques (Wiley & Sons, New York, New York, 2002).
3. J.W. Jaroszewski, Planta Med., 71(8), 691–700 (2005).
4. J.W. Jaroszewski, Planta Med., 71(9), 795–802 (2005).
5. O. Corcoran and M. Spraul, Drug Discov. Today, 8(14), 624–631 (2003).
6. L. Griffiths and R. Horton, Magn. Reson. Chem., 36(2), 104–109 (1998).
7. N.T. Nyberg, H. Baumann and L. Kenne, Magn. Reson. Chem., 39(5), 236–240 (2001).
8. N.T. Nyberg, H. Baumann and L. Kenne, Anal. Chem., 75(2), 268–274 (2003).
9. S.R. Wilson et al., J. Sep. Sci., 30(3), 322–328 (2007).
10. F. Xu and A. Alexander, Magn. Reson. Chem., 43(10), 776–782 (2005).
11. A. Alexander, F. Xu and C. Bernand, Magn. Reson. Chem., 44(1),1–6 (2006).
12. G. Miliauskas et al., J. Nat. Prod., 68(2), 168–172 (2005).
13. D. Sorensen et al., J. Nat. Prod., 70(1), 121–123 (2007).
14. M. Godejohann et al., J. Chromatogr. A, 1058(1–2), 191–194 (2004).
15. S.R. Wilson et al., J. Sep. Sci., 29(4), 582–589 (2006).
16. M.E. Lacey, et al., Chem. Rev., 99(10), 3133–3152 (1999).
17. D.L. Olson et al., Anal. Chem., 76(10), 2966–2974 (2004).
18. K. Putzbach, et al., J. Pharm. Biomed. Anal., 38(5), 910–917 (2005).
19. M. Krucker et al., Anal. Chem., 76(9), 2623–2628 (2004).
20. M. Gronquist et al., J. Am. Chem. Soc., 127(31), 10810–10811 (2005).
21. J.F. Hu et. al., Planta Med., 71(2), 176–180 (2005).
22. J.F. Hu et. al., Phytochem. Anal., 16(2), 127–133 (2005).
23. E.M. Thurman and M.S. Mills, Solid-Phase Extraction (Wiley & Sons, New York, 1998).
24. S. Bieri, et al., Phytochem. Anal., 17(2), 78–86 (2006).
25. G. Miliauskas et al., J. Chromatogr. A, 1112(1–2), 1276–284 (2006).
26. M. Sandvoss et al., Magn. Reson. Chem., 43(9), 762–770 (2005).
27. M. Lambert et al., Magn. Reson. Chem., 43(9), 771–775 (2005).
28. M. Lambert et al., J. Nat. Prod., 68(10), 1500–1509 (2005).
29. C. Seger et al., Anal. Chem., 77(3), 878–885 (2005).
30. V. Exarchou et al,. Anal. Chem., 75(22), 6288–6294 (2003).
31. S. Christophoridou et al., J. Argi. Food Chem., 53(12), 4667–4679 (2005).
32. V. Exarchou et al., J. Chromatogr. A, 1112(1–2), 293–302 (2006).
33. C. Clarkson et al., J. Nat. Prod., 69(4), 527–530 (2006).
34. C. Clarkson et al., J. Nat. Prod., 69(9), 1280–1288 (2006).
35. E.C. Tatsis et al., Phytochemistry, 68(3), 383–393 (2007).
36. S.H. Lam et al., Phytochem. Anal., 18(3), 251–255 (2007).
37. A. Pukalskas, T.A. van Beek and P.de Waard, J. Chromatogr. A, 1074(1–2), 81–88 (2005).
38. M. Lambert et al., Anal. Chem., 79(2), 727–735 (2007).
39. C. Clarkson et al., Planta Med. 73(6), 578–584 (2007).
40. C. Clarkson et al., Anal. Chem., 77(11), 3547–3553 (2005).
41. C. Seger et al., J. Chromatogr. A, 1136(1), 82–88 (2006).
42. C.Y. Wang and S.S. Lee, Phytochem. Anal., 16(2), 120–126 (2005).
43. S.S. Lee et al., J. Nat. Prod., 70(4), 637–642 (2007).
44. C. Seger et al., J. Chromatogr. A, 1163(1–2) 138–144 (2007).
45. A.J. Simpson et al., Analyst, 129(12), 1216–1222 (2004).
46. A.W. Nicholls et al., Xenobiotica, 36 (7), 615–629 (2006).
47. C. Pan et al., J. Pharm. Biomed. Anal., 40(3), 581–590 (2006).
48. N.P. Vasilev et al., J. Biotechnol., 126(3), 383–393 (2006).


















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



