HPLC for Characterization and Quality Control of Therapeutic Monoclonal Antibodies
LCGC North America


The latest approaches to antibody characterization go beyond ion-exchange, size-exclusion, and reversed-phase modes to make use of affinity, hydrophobic interaction, mixed-mode, and HILIC techniques.
Monoclonal antibodies (mAbs) are highly complex recombinant proteins used successfully as therapeutics in many disease indications. Myriad analytical techniques are required to characterize and control the quality of these molecules during their clinical development and commercial production. This installment describes recent advances and best practices in high performance liquid chromatography (HPLC) analysis of intact mAbs and their variants and fragments. Application examples, supported by key references, are included to illustrate the utility of diversified HPLC modes to characterize these heterogeneous molecules.
Monoclonal antibodies (mAbs) are established therapeutics with multiple blockbuster drugs in oncology and inflammatory diseases (1–3). According to Genetic Engineering & Biotechnology News published on-line on March, 3, 2014, there were five antibody drugs (Humira, Remicade, Rituxan, Avastin, and Herceptin) and one fusion protein (Enbrel) among the top 10 drugs in 2013, and these accounted for nearly $50 billion in global sales. Hundreds of mAbs are currently under clinical development, targeting diseases in autoimmunity, inflammation, cancer, infection, ophthalmology, and other indications (4). Beyond traditional mAb formats, bispecific mAbs and antibody-drug conjugates (ADCs) are major platforms of active research and development (4–7).


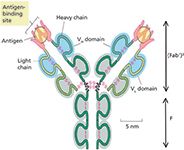
Figure 1: Structure of a typical antibody molecule with two identical heavy chains and light chains and antigen-binding sites. VH and VL are the variable domains in the heavy and light chains. The molecules can be cleaved enzymatically into a lower Fc and two Fab fragments. Adapted with permission from reference 8.
MAbs are immunoglobulins (150 kDa) with a typical structure shown in Figure 1 (8). Important structural characteristics and physicochemical properties of mAbs are summarized in Table I and have been reviewed (9,10). Biological activity (that is, therapeutic efficacy) of the protein depends on the exact three-dimensional (3D) structure, which is likely affected by post-translational modifications or degradations (11). In contrast with small-molecule drugs, which are chemically synthesized by a variety of processing routes to achieve a high purity product, recombinant human mAbs (rhMAbs) are derived from a fermentation process using host cells such as Chinese hamster ovarian (CHO) cells, followed by extensive purification. Because of their inherent heterogeneity, the quality of mAb therapeutics is controlled by the production cell line and the bioprocessing parameters used for each batch (7,12,13).


Table I: Important structural characteristics and physicochemical properties of mAbs
Critical quality attributes (CQAs) of biopharmaceuticals are described by the International Conference on Harmonization Guideline Q8 (R2). CQAs are a set of physical, chemical, biological, or microbiological properties or characteristics that should be kept within an appropriate limit, range, or distribution to ensure the desired product quality, safety, and efficacy. CQAs are monitored closely during each bioprocessing step, and can be incorporated into the release specifications for drug substances and drug products as exemplified by Table II (14). Important characteristics such as identity, purity, strength, potency, composition, and safety are obtained from multiple complementary analytical techniques, including various modes of high performance liquid chromatography (HPLC).

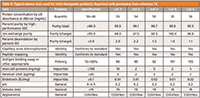
Table II: Typical release tests used for mAb therapeutic products. Reprinted with permission from reference 14.
Principles of quality risk management and assessment and quality by design (QbD) serve as the foundation for current regulatory expectations for process development (15,16). As an integral part of QbD, assessment of CQAs could be based on their impacts in four categories: bioactivity, pharmacokinetics–pharmacodynamics (PK-PD), immunogenicity, and safety. Typical mAb CQAs include size, charge, oxidation, aggregation, glycosylation, and cysteine variants. Other CQAs include process impurities such as residual levels of host-cell DNAs and proteins, antibiotics, as well as contaminants such as mycoplasma, viruses, or proteases (13,15).
Analytical methodologies used for quality control (QC) and characterization are the cornerstones of mAb drug development processes. A well-designed quality control system includes procedure controls (in-process monitoring or controls), raw material or excipient controls, characterization testing, stability testing, and final lot release testing and shipping. Regulatory guidances define the requirements of QC practices, which are applied to characterization and method validation (17). An effective QC strategy should provide assurance that products are safe, have consistent quality, and include standard procedures to guard against unexpected incidents that impact drug quality. In the end, the QC-released mAbs must meet predefined specifications acceptable to regulatory agencies (14).
To monitor the CQAs of heterogeneous mAb products, multiple analytical techniques are used. These include chromatography (HPLC), electrophoresis (capillary gel electrophoresis, capillary isoelectric focusing, sodium dodecyl sulfate polyacrylamide gel electrophoresis [SDS PAGE]), spectroscopy (mass spectrometry [MS], UV–vis spectrophotometry, nuclear magnetic resonance [NMR], infrared [IR] spectroscopy, circular dichroism), and other techniques (multiple-angle laser scattering, differential scanning calorimetry, enzyme-linked immunosorbent assay [ELISA], microbial testing, sterility testing, and binding and functional bioassays) (13,14,18,19). The use of HPLC (11,20–23) and ultrahigh-pressure liquid chromatography (UHPLC) (24–30) for the analysis of proteins and peptides has been reviewed in many books and journal articles (11,20,22–24,26,30,31). In this installment, we focus our discussion on the various modes of HPLC used for extended characterization, lot release testing, and stability testing of intact therapeutic mAbs and their variant and degradant forms. Selected examples and reviews of key references are used to illustrate the best practices and recent developments in mAb analyses.
Size-Exclusion Chromatography
Size-exclusion chromatography (SEC) separates molecules based on molecule size by selectively excluding them from an inert porous matrix of controlled pore sizes (32). SEC is typically performed under nondenaturing operating conditions at room temperatures with isocratic mobile phases containing 0.2 M to 0.5 M sodium chloride or potassium chloride and buffered to approximately pH 6. Typical stationary phases are silica- or polymer-based supports with a hydrophilic coating or bonded layer. The Tosoh TSKgel SEC column is widely used in the biopharmaceutical industry for mAbs analyses and has been on the market since 1987 (33).
The recent introduction of ultrahigh-performance SEC columns packed with small particles provides an opportunity for shortened analysis time and better peak resolution. For example, Jeong and colleagues (31) demonstrated a 5-min separation of an Fab dimer and monomer on a 1.7-µm bridged-ethyl hybrid (BEH) column with improved peak resolution in comparison to an existing 30-min standard HPLC separation (31). This fast SEC method has been qualified for robustness, accuracy, and precision. Diederich and colleagues (34) demonstrated that an interlaced injection technique that could further speed up the decreased SEC analysis time. By comparison of three SEC columns (TSKgel 3000 SWxl, Zenix SEC-250 [Sepax Technologies], and Acquity UPLC BEH200 [Waters]) in terms of throughput, resolution, and precision, they indicated that similar profiles were obtained (Figure 2) and that the Acquity UPLC BEH200 column has the highest resolution, whereas the Zenix column offers an economical alternative to the TSKgel column. This UHPLC method has a 6-min cycle time for single injections versus <2-min cycle time in the interlace format (staggered injections).


Figure 2: (a) Overlay of single-injection chromatograms of the mAb sample (1.0 g/L) analyzed on three different SEC columns. (b) For comparability, elution volumes were normalized to column void volumes. Columns: 150 mm à 4.6 mm, 200-à 1.7-µm Acquity UPLC BEH200 SEC: 250 mm à 4.6 mm, 300-à 3-µm Zenix SEC-250; 300 mm à 7.8 mm, 250-à 5-µm TSKgel 3000 SWxl. Mobile phase: 0.2 M potassium phosphate buffer at pH 6.2, containing 0.25 M potassium chloride. Adapted with permission from reference 34.
High-molecular-weight species (HMWS) such as aggregates in mAbs have been correlated with immunogenicity, a high-impact safety risk (35). SEC is a key technique to monitor HMWS as well as other low-molecular-weight species (LMWS). Arakawa and colleagues (36) identified several caveats when implementing SEC, such as its low resolution and the potential of nonspecific adsorption on the column, which leads to inaccurate molecular mass estimation. A careful selection of SEC method parameters is therefore needed to obtain useful data that correlate well with results from analytical ultracentrifugation, multiple-angle light scattering (MALS), or MS.
In a critical evaluation of ultrahigh-performance SEC separations for mAbs aggregates, Fekete and colleagues (24)observed high plate counts using sub-2-µm columns, but comparatively higher HMWS values of 7–10% versus approximately 4% with conventional HPLC. Although routine SEC analysis does not require high temperatures, these results suggested the possibility of aggregation artifacts formed during the UHPLC analysis at high temperatures and pressure. Our laboratories are conducting further investigation of this aspect of ultrahigh-performance SEC, and results will be reported elsewhere.
Ion-Exchange Chromatography
Charge-based separation analyses are typically included in the release and stability specifications for QC and comparability studies to support post-approval process changes. Ion-exchange chromatography (IEC) is a key purity assay for mAb charge variants, that are often functionally active species (37). Polymer-based columns, the most widely used for IEC analysis, are coated with a hydrophilic layer functionalized with ion-exchange groups such as carboxylate, sulfonate, amino, and quaternary amine, and elution is accomplished with salt or pH gradients. Although their numbers are still limited, the increasing availability of sub-3- and sub-2-µm IEC columns is beginning to impact the performance of these separations (38).
Approximately 20% of the amino acid residues in a typical human IgG1 platform molecule are charged in nature. Post-translational modifications or degradations of these charged residues create variant forms, where either a charged site becomes removed (that is, Lys epsilon amine loss by glycation, amino-terminal free primary amine loss by cyclization, carboxyl-terminal Lys cleaved), or a new charged site is produced (for example, Asn deamidation into Asp, glycan sialylation). Alterations in charge-based content of mAb variants impact interaction with IEC ligands, resulting in different retention profiles. An IEC charge variant method can be used analytically for monitoring changes to the product profile, or preparatively as an isolation technique of new variants for further characterization or identification.
An IEC charge variant profile for mAbs is typically generated from a negatively charged carboxylate or sulfonate cation-exchange column. Note that immunoglobulin G (IgG) molecules generally are basic in nature, with an isoelectric point at pI > 7 in a near-neutral pH buffer mobile phase system. An ion-exchange profile ideally presents a predominant main peak with minor flanking regions of acidic and basic variants eluting before or after the main peak, respectively (39). IEC profiles of mAb charge variant forms are typically heterogeneous because of multiple modifications on many sites throughout the molecule, often resulting in a series of unresolved peaks, particularly on the acidic side. In contrast, modifications to the charged termini residues are often well resolved from each other and the main peak.
A comprehensive overview of charge variant modifications and their impact to IEC profile shifts with an extended focus on deamidation and isomerization degradation were documented by Vlasak and Ionescu (40). In a 2001 paper, Harris and colleagues (19) demonstrated the IEC separation, isolation, and subsequent characterization for six different charge variants of the rhuMAb HER2 IgG1 molecule. Strikingly well resolved variants, which differed from the main-peak intact IgG1 native form by a single Asn deamidation (negative charge added, material shifted to acidic) or Asp isomerization (charge preserved, material shifted to basic), illustrated the power of an IEC separation. Chumsae and colleagues (41) profiled a typical mAb IEC separation from which an unusual arginine modification by methylglyoxal was identified by an acidic shift. Removal of positive charges resulted in molecules that were significantly shifted into the acidic region, and seven charge-modified arginine residues, which were located in accessible, flexible complementarity determining region (CDR) sequences, were identified from isolated fractions.
A frequently used strategy to address the poorly resolved charged variants utilizes a digestion of the mAb molecule with papain before IEC profiling. Creation of smaller Fab and Fc portions reduces the complexity generated by multiple modifications across the intact heterodimeric molecule, and often results in cleaner profiles with more distinct peaks. An interesting example from Moorhouse and colleagues (42) illustrated this approach, where Fc C-terminal lysine variants can be separately assessed from Fab amino-terminal glutamine cyclized variant forms. Individual terminal species with charge differences were fully resolved and sequentially eluted in order of increasing pI: Fc + 0 C-terminal K, Fc + 1K, Fc + 2K, Fab + 2 N-terminal pyroglutamates (pE), Fab + 1pE + 1 glutamine (Q), Fab + 2Q.
Pace and colleagues (43) applied this same fragmentation IEC strategy to generate independent Fab and Fc deamidation rates in stability studies to assess and improve product shelf life. Kinetic degradation rates were determined based on the level of acidic charge variants in each fragment in response to varying buffer components, pH, and column temperature. Susceptible deamidation residues in the CDR-containing FAb portion could thus be specifically stabilized to extend shelf life. Zhang and colleagues (44) used IEC to monitor a previously unidentified acidic variant form that occurs in the Fc domain under thermal stress conditions (Figure 3). Characterization analyses of the acidic modification identified Asn-330 as an unexpected site of deamidation and isomerization, in the Val-Ser-Asn-Lys sequence. IEC appears to be able to effectively monitor Fc Asn-330 deamidation and isomerization, a major degradation pathway under thermal stress conditions.
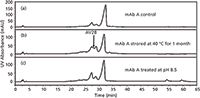
Figure 3: Cation-exchange analysis of mAb A samples under various conditions: (a) unstressed control, (b) thermally stressed at 40 °C for four weeks at pH 5.3, and (c) mAb A treated with pH 8.5. The arrow in (b) points to a new degraded peak unique to thermally stressed samples and termed "AV2B" in the paper. Adapted with permission from reference 44.
Reversed-Phase Liquid Chromatography
Reversed-phase liquid chromatography (LC) analysis of mAbs has multiple applications across all structural levels, from full-sized intact molecules to Fab and Fc fragment molecules and reduced heavy- and light-chain species, and peptide maps generated by proteolytic digestion (11,21,28,45). The broad utility of reversed-phase LC is because of its well-studied hydrophobic separation mechanism, the availability of very efficient reversed-phase LC columns packed with small-particle, nonporous, fully porous, or superficially porous (SPP) materials, and the use of mobile phases compatible with MS for peak identification and structure elucidation (20,45,46).
Many researchers have used reversed-phase LC to exploit these hydrophobic differences in mAb variant forms at the intact or large-fragment levels to provide a quick assessment of process and formulation development samples. Identified variant forms that exhibited altered hydrophobicity (which controls reversed-phase LC retention) include methionine oxidation, glycosylation, disulfide-free thiol status, and amino-terminal cyclization. Peptide mapping by reversed-phase LC–MS is the basis for de novo sequencing, identity assay for lot release testing, and detailed characterization or identification of variant forms (45).
Reversed-phase LC of proteins and peptides has been an area of intensive research for several decades. The fundamentals and separation mechanism are generally understood and well-documented (11,20,23). Recent column developments specifically for mAbs have used approaches such as bonded phases with low silanophilic silica for better peak shape, short alkyl chain length ligands to reduce retention and adsorption, nonporous, superficially porous, or wide-pore materials to improve mass transfer, and sub-3-µm or sub-2-µm particles for enhanced efficiencies (20,23,26,31,46).
Fekete and colleagues (47) explored the impact of mobile phase and column temperature on two new wide-pore packing materials (1.7-µm fully porous and 3.6-µm core–shell particles) on the stability and recovery of commercially manufactured IgG molecules (intact IgGs and fragments: heavy chain, Fc, and Fab). To mitigate strong secondary ionic interactions, the authors operated the column at high temperatures (80–90 °C), along with incorporating small amounts (1–3%) of a protic solvent in the mobile phase (such as n-butanol) to enhance peak shape and recovery. In a separate paper, the same research group demonstrated the use of a coupled-column approach to enhance resolution of mAb separations (30).
An earlier study published by Dillon and colleagues (48) described the use of reversed-phase LC–MS to screen intact mAb for heterogeneity associated with post-translational modifications (that is, glycosylation) and stability degradations (clipped species). In addition, conformational isoforms were detected and attributed to disulfide linkage heterogeneity for an IgG2 (49). Rehder and colleagues (50) applied similar optimization of reversed-phase LC–MS methods to analyze reduced and alkylated IgG light- and heavy-chain (LC and HC) molecules and for monitoring intact glycan distribution (G0, G1, G2, Man5) and amino-terminal cyclizations.
Reversed-phase LC analysis of intact mAb also provided an unexpected separation of free thiol variant forms as demonstrated by Zhang and colleagues (51). Under the denaturing conditions of the reversed-phase LC environment (high temperature, low pH, and organic solvent), variants containing unpaired intrachain disulfides were completely separated from the expected, disulfide-bonded paired cysteine mAb (Figure 4). Free thiol variant forms presented as more hydrophobic than completely disulfide-bonded material, and were observed for both the intact mAb and the FAb fragment. The separated minor peaks, which could be completely converted back into the main peak by CuSO4 treatment, were characterized as the unbonded free thiol form of the Cys22–Cys96 disulfide pair located in the VH region.


Figure 4: Reversed-phase LC profiles for the intact mAb A with buried unpaired cysteines using three different columns: (a) 150 mm à 4.6 mm, 3-µm Agilent (Varian) diphenyl; (b) Zorbax SB-C8; and (c) Agilent (Varian) PLRP-S column; mobile-phase A: 0.1% trifluoroacetic acid in water; mobile-phase B: 0.1% trifluoroacetic acid in acetonitrile; gradient: 36â45% B in 18 min; flow rate: 1 mL/min; temperature: 75 °C; detection: UV absorbance at 280 nm. Adapted with permission from reference 51.
Affinity Chromatography
Affinity chromatography (52) utilizes a highly specific, often biologically based binding interaction to capture specific molecules. Antibody molecules themselves frequently serve as highly selective immobilized ligands for isolation of analytes of interest from complex matrices. Common stationary phases in affinity chromatography are protein A or protein G for IgG and boronate affinity for glycated proteins. Affinity chromatography is routinely used during the purification process for therapeutic mAb production. IgG affinity interactions with protein A or protein G are exploited both at large-scale for an initial high-capacity chromatographic separation of the product from host cell materials, and at small-scale for fast, high-throughput analysis for product titer (12). This high-affinity interaction occurs within the Fc CH2–CH3 regions, and after extraneous, nonretained host cell materials are washed through. Isolated IgG is typically eluted with an acidic pH step-gradient wash.
Interestingly, affinity chromatography has been used to further separate Fc region oxidation variants by applying linear gradients for elution. Loew and colleagues (53) explored the impact of oxidation at two highly conserved methionine residues, Met-252 and Met-428, which are located in the Fc CH2–CH3 regions, and are directly involved in the binding interaction with protein A. Schlothauer and colleagues (54) exploited IgG affinity for the neonatal Fc receptor protein (FcRn), a physiologically functional interaction with Fc CH2–CH3 regions. FcRn affinity columns, prepared in-laboratory via biotinylated receptor material coupled to streptavidin-sepharose, were operated under physiological pH gradient conditions to elute bound IgG or Fc material.
Another important application of affinity chromatography is boronate chromatography for antibody glycation characterization. Quan and colleagues (55) used an optimized separation that contained hydroxylated shielding reagents in the mobile phase when assaying basic IgG molecules. Shielded boronate affinity chromatography can be effectively used to quantify, isolate, and characterize glycated IgG1.
Hydrophobic Interaction Chromatography
Hydrophobic interaction chromatography (HIC) (56) is a separation mode in which the molecules in high-salt mobile phase environments interact hydrophobically with nonpolar stationary phases (for example, hydrophobic ligand bonded to a polymer or a silica support with a hydrophilic outer layer). Common mobile phases are 1–2 M ammonium sulfate buffered at ~pH 6. HIC is an "orthogonal" technique (with very different selectivity) to reversed-phase LC, IEC, or SEC and is useful for characterization of intact and large fragment IgG molecules (56). HIC uses an initial matrix of high-concentration water-structuring salt to expose IgG hydrophobic regions, which can then interact with an alkyl- or phenyl-functionalized stationary phase. A decreasing salt concentration gradient is used to elute the various IgG forms based on relative hydrophobicity. HIC is generally applicable for initial broad fractionation of complex protein solutions (for example, whole cell broths and plasma). However, analytical scientists can apply this technique to effect separations on highly purified IgG product with very subtle modifications. HIC is also a reference technique for ADC drugs for the determination of antibody–drug ratio (57).
HIC methodologies have provided very effective separations for monitoring mAb aspartic acid isomerization and thiol variants. Cacia and colleagues (58) used an analytical TSK-Butyl (Tosoh) column to separate seven hydrophobically distinct species, related to aspartic acid isomerization variants of an intact IgG1 in a 14-min method. Wakankar and colleagues (59) further utilized an optimized HIC method to generate stability data and degradation rates for Fab aspartic acid isomerization on the same IgG1 molecule. The resulting profile had additional hydrophobic species separated, which were identified as the Fab intact intrachain disulfide and free thiol forms at heavy chain Cys22–Cys29. Harris and colleagues (18) documented the final HIC separation of six Fab-associated peaks: in order of increasing retention and hydrophobicity, LC-Asp32/HC-Cys22-Cys96 disulfide (native material), LC-Asp32/HC-Cys22-Cys96 free thiols, LC-isoAsp32/HC-Cys22-Cys96 disulfide, LC-isoAsp32/HC-Cys22-Cys96 free thiols, LC-Succinimide32/HC-Cys22-Cys96 disulfide, LC-succinimide32/HC-Cys22-Cys96 free thiols.
Likewise, Dick and colleagues (60) used HIC analysis of an intact mAb for accelerated stability studies by temperature-induced degradation. Isomerization at a heavy-chain CDR2 aspartic acid was quantitatively monitored to establish degradation rates and determine the impact of formulation buffer components. Valliere-Douglass and colleagues (61) used a modified HIC methodology to improve recovery of an IgG2 molecule used in stability studies, by adding acetonitrile to a low pH (5.2) mobile phase and replacing ammonium sulfate with ammonium acetate as the elution salt.
HIC is an important method to determine an overall drug-to-antibody ratio. Wakankar and colleagues used HIC with low concentrations of acetonitrile added to the low-salt mobile phase to help ensure full elution of antibody-drug conjugates (ADCs) loaded with higher-order drug levels. Using a butyl-nonporous stationary phase, five species of an intact IgG linked with an increasing number of a small-molecule drugs (zero, two, four, six, and eight) were baseline resolved in less than 15 min (Figure 5) (57).

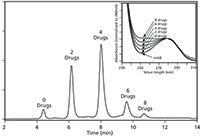
Figure 5: Hydrophobic interaction chromatography (HIC) analysis of a mAb-vc-MMAE on a Tosoh Bioscience Butyl-NPR column yields five predominant peaks that correspond to mAb containing zero, two, four, six, and eight small-molecule drugs. Inset: An overlay of the UV spectra of the starting mAb and the HIC peaks, normalized to the 280 nm absorbance, showing the increase in the 248 nm absorbance as the level of conjugated linker-drugs increases. Adapted with permission from reference 57.
The wide applications of HIC for antibody analysis were further demonstrated by Valliere-Douglass and colleagues (62), who developed a variety of finely tuned, optimized HIC separations for intact IgG, Fab, and Fc fragment species. Multiple aspartate isomerization-based intermediates and isoforms in CDRs were fully separated for different IgG molecules. After peaks were characterized, HIC analysis proved to be a superior monitoring method for these degradation changes, as well as for isolating material for functional assessment. Fc fragment variant species were also well separated, including oxidized methionine forms and processed C-terminal forms such as lysine removal and proline amidation. Finally, HIC provided an efficient separation of size-based variant forms, particularly for isolating and enriching clipped species for further characterization.
Mixed-Mode Chromatography
Mixed-mode chromatography uses multiple forms of interactions between analytes and the stationary phase (for example, IEC, HIC, SEC) (63). Mixed-mode chromatography offers unique selectivity for some variants that are difficult to separate by other modes of chromatography, and has been successfully utilized for large-scale protein purification in biopharmaceutical manufacturing (63). Unlike reversed-phase LC that denatures proteins, mixed-mode chromatography can be applied to isolate mAbs for further biological characterization.
The mixed-mode phenomenon in SEC is a nuance that has been exploited for the separation of oxidized antibodies. By coupling two TSK-Gel (Tosoh) columns in series, Yang and colleagues (64) observed a pre-peak in SEC. Further characterization of the pre-peak revealed the oxidation of trypotophan residues (Trp-103) on the heavy chain of the IgG2. Wong and colleagues (65) exploited to separate oxidation variants by running ultrahigh-performance SEC columns under moderate HIC conditions, which separated an IgG2 into size-related variants, and additionally resolved a monomer pre-peak from the monomer main peak. The pre-peak was confirmed by orthogonal characterization techniques as an oxidized Trp-104 IgG monomer, which was 40–50% less active because of the location of Trp-104 in the CDR. The IgG tryptophan oxidation that was generated under the stressed condition can be clearly monitored by this mixed-mode method.
Hydrophilic-Interaction Chromatography
Hydrophilic-interaction chromatography (HILIC) (56,66,67) provides enhanced retention and selectivity for separations of polar components. The stationary phases used include bare silica, and silica bonded with amide, amine, cyano, and diol polar functional groups. More-complex silica derivatizations produce anion-exchange, cation-exchange, zwitterionic, and mixed-mode stationary phases that can be operated in HILIC mode with appropriate mobile phases. Typical mobile phases are initial high concentrations of polar organic solvents (mostly aprotic but sometimes protic) that are water-miscible (for example, acetonitrile, alcohols) with aqueous buffers for pH and ionic strength control.
HILIC has been applied to mAb carbohydrate analysis, separating these highly polar glycans compounds in a MS-compatible mobile phase. Ahn and colleagues (68) developed a HILIC separation of fluorescently labeled N-linked glycan structures released from human IgG by using UHPLC. Baseline separation was achieved within a reasonable time (<1 h) for the expected neutral and charged glycans, including many structural isomers.
Peptide mapping by HILIC provides complementary information to typical reversed-phase LC maps. Zhu and colleagues (69) applied HILIC peptide mapping with unbonded silica to a highly glycosylated therapeutic protein (erythropoietin [EPO]). Although EPO is not a mAb, this work demonstrated the general applicability of orthogonal selectivity, both for very hydrophilic peptides that are not well retained by reversed-phase LC.
The use of HILIC for intact protein analysis remains limited, primarily because of protein incompatibility issues in the initial mobile phase. Many intact proteins including IgGs, become either denatured or are not soluble in initial conditions. Denatured intact proteins are often not recovered in an active form, or remain irreversibly adsorbed to the HILIC stationary phase. Tetaz and colleagues (66) studied HILIC applications for a set of intact, nonaqueous soluble proteins that are lipophilic or hydrophobic membrane-associated in nature, and demonstrated glycosylation-based, isoform separations of intact proteins.
Challenges and Opportunities for Future Development
The emerging antibody formats such as ADCs have created new challenges for separation scientists. The complexity of an antibody coupled with a linker and a cytotoxic drug creates greater heterogeneity in an ADC molecule, and typically does not yield discrete peaks in traditional IEC for mAbs with and without the bound drugs (57). Similarly, bispecific antibodies based on molecules with two different Fabs containing two distinctive binding sites for two different antigens, comprise another platform under intense development (5). The production and assembly of bispecific mAbs are more demanding on separation techniques for greater resolution of numerous variants of the desired forms.
To address the increasing need for higher resolution and selectivity in mAbs characterization, academic research labs have developed new materials that perform beyond the traditional limitations for large molecule chromatography. A notable example is an ultra-efficient chromatography based on submicrometer colloidal silica particles developed by Professor Mary Wirth's group at Purdue University (70). They demonstrated that a novel "slip flow" phenomenon could enable a very high efficiency separation of antibody aggregates in 40 s (Figure 6). The separation can be carried out at room temperature in contrast to typical reversed-phase-UHPLC conducted at higher column temperatures (70–80 °C), avoiding potential on-column degradation.

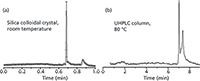
Figure 6: Separation of a labeled monoclonal antiprostate specific antigen and its aggregate using "slip flow" ultra-efficient chromatography: (a) Silica colloidal crystal: 21 mm à 0.075 mm capillary column packed with 0.47 µm colloidal silica bonded with C4 ligand; mobile phase: 40% acetonitrile in 0.1% trifluoroacetic acid; temperature: ambient. (b) Commercial column, 50 mm à 2.1 mm column packed with 1.7-µm C4 material; temperature: 80 °C. Adapted with permission from reference 71.
Multidimensional LC has been successful for increasing peak capacity for complex samples and is extensively used in proteomics research (71). For mAbs characterization, multidimensional LC techniques can reveal variants undetected in just one mode of separation and provide more in-depth understanding of the molecules. Since minor variants are present at low abundance and often are eluted near other peaks, an effective enrichment technique such as displacement chromatography could be initially applied (39), coupled with a second dimension to achieve better resolution of minor components.
Summary and Conclusion
Monoclonal antibody therapeutics require a wide array of HPLC modes for characterization and quality control applications where CQAs such as charge, size, oxidation, cysteine forms, and glycan variants are monitored in process development. These complementary techniques are used to reveal minor changes to the mAb molecules or for the determination of low levels of aggregates, variants, and impurities. Judicious applications of these HPLC separation modes are vital to a more thorough understanding of these complex biopharmaceuticals and their associated manufacturing processes that can impact final product quality. Robust and high-performance HPLC techniques are the cornerstones for mAb development and production, pivotal in assuring the safety and efficacy of these targeted therapeutics for many serious diseases.
Acknowledgments
The authors are grateful to many colleagues for their useful inputs and suggestions, including Drs. Yung-Hsiang Kao, John Stults, and Reed Harris, and Yi Li of Genentech; Drs. Davy Guillarme and Szabolcs Fekete of University of Geneva; Professor Mary Wirth of Purdue University; Professor Anurag Rathore of Indian Institute of Technology; Chris Pohl of Thermo Scientific; John Batts from IDEX Health & Science; Dr. Tom Waeghe from MacMod; Dr. Di Gao of MedImmune; and C. David Carr of Bioanalytical Technologies. The opinions expressed in this article are solely those of the authors and bear no reflections on those from LCGC North America or other organizations.
References
(1) P.J. Carter, Nat. Rev. Immunol. 6(5). 343–357 (2006).
(2) Z. An, Therapeutic Monoclonal Antibodies: From Bench to Clinic (John Wiley & Sons, Hoboken, New Jersey, 2011).
(3) G. Walsh, Biopharmaceuticals: Biochemistry and Biotechnology (John Wiley & Sons, Hoboken, New Jersey, 2006).
(4) J.M. Reichert, mAbs 6(1), 5–14 (2014).
(5) A. Beck, T. Wurch, C. Bailly, and N. Corvaia, Nat. Rev. Immunol. 10(5), 345–352 (2010).
(6) P.J. Carter and P.D. Senter, The Cancer Journal 14(3), 154–169 (2008).
(7) J.M. Reichert and V.E. Valge-Archer, Nat. Rev. Drug Discovery 6(5), 349–356 (2007).
(8) B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts, and P. Walter, Molecular Biology of the Cell, 5th edition (Garland Science. New York, New York, 2008) pp. 157.
(9) A. Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer, and S. Sanglier-Cianférani, Anal. Chem. 85(2), 715–736 (2012).
(10) H. Liu, G. Gaza-Bulseco, D. Faldu, C. Chumsae, and J. Sun, J. Pharm. Sci. 97(7), 2426–2447 (2008).
(11) C.D. Carr, LCGC North Am. 32(s4), 24–29 (2014).
(12) S.J. Shire, W. Gombotz, K. Bechtold-Peters, and J. Andya, Current Trends in Monoclonal Antibody Development and Manufacturing (Springer, Boca Raton, Florida, 2010).
(13) K.B. Seamon, Curr. Opin. Biotechnol. 9(3), 319–325 (1998).
(14) A.S. Rathore, BioPharm Int. 23(1), 46–53 (2010).
(15) A. Eon-Duval, H. Broly, and R. Gleixner, Biotechnol. Prog. 28(3), 608–622 (2012).
(16) P. van Hoek, J. Harms, X. Wang, and A.S. Rathore in Quality by Design for Biopharmaceuticals: Principles and Case studies, A.S. Rathore and R. Mhatre, Eds. (John Wiley & Sons. Hoboken, New Jersey, 2009), pp. 85–109.
(17) J. Geigert, The Challenge of CMC Regulatory Compliance for Biopharmaceuticals and Other Biologics (Springer, New York, New York, 2013).
(18) R.J. Harris, E.T. Chin, F. Macchi, R.G. Keck, B.-J. Shyong, V.T. Ling, A.J. Cordoba, M. Marian, D. Sinclair, and J.E. Battersby, Current Trends in Monoclonal Antibody Development and Manufacturing (Springer, 2010), pp. 193–205.
(19) R.J. Harris, B. Kabakoff, F.D. Macchi, F.J. Shen, M. Kwong, J.D. Andya, S.J. Shire, N. Bjork, K. Totpal, and A.B. Chen, J. Chromatogr. B: Biomed. Sci. Appl. 752(2), 233–245 (2001).
(20) M. Dong, J. Gant, and B. Larsen, BioChromatography 4(1), 19–34 (1989).
(21) M.W. Dong, Modern HPLC for Practicing Scientists (Wiley, Hoboken, New Jersey, 2006).
(22) K.M. Gooding and F.E. Regnier, HPLC Of Biological Macro-Molecules, Revised And Expanded (CRC Press, Boca Raton, Florida, 2002).
(23) R.L. Cunico, K.M. Gooding, and T. Wehr, Basic HPLC and CE of Biomolecules (Bay Bioanalytical Laboratory, Richmond, California, 1998).
(24) S. Fekete, K. Ganzler, and D. Guillarme, J. Pharm. Biomed. Anal. 78–79, 141–149 (2013).
(25) J. Jeong, T. Zhang, J. Zhang, and Y.H. Kao, Amer. Pharm. Rev. 14(2), 44–51 (2011).
(26) J.C. Rea, Y.J. Wang, and T. Zhang in Ultra-High Performance Liquid Chromatography and Its Applications, Q.A. Xu, Ed. (Wiley. Hoboken, New Jersey, 2013), pp. 235–252.
(27) D. Guillarme and J.-L. Veuthey, UHPLC in Life Sciences (Royal Society of Chemistry, Cambridge, UK, 2012).
(28) I. Krull and A. Rathore, LCGC North Am. 28(6), 454–468 (2010).
(29) I. Krull, A. Rathore, and T.E. Wheat, LCGC North Am. 29(12), 1052–1062 (2011).
(30) S. Fekete, M. Dong, W., T. Zhang, and D. Guillarme, J. Pharm. Biomed. Anal. 83, 273–8 (2013).
(31) J. Jeong, T. Zhang, J. Zhang, and Y.-H. Kao, Am. Pharm. Rev. 14(2), 44 (2011).
(32) C.-S. Wu, Handbook Of Size Exclusion Chromatography and Related Techniques: Revised And Expanded (CRC Press, Boca Raton, Florida, 2003).
(33) Y. Kato, Y. Yamasaki, H. Moriyama, K. Tokunaga, and T. Hashimoto, J. Chromatogr. A 404, 333–339 (1987).
(34) P. Diederich, S.K. Hansen, S.A. Oelmeier, B. Stolzenberger, and J. Hubbuch, J. Chromatogr. A 1218(50), 9010–9018 (2011).
(35) A. Mire-Sluis, B. Cherney, R. Madsen, A. Polozova, A. Rosenberg, H. Smith, T. Arora, and L. Narhi, BioProcess Int. 9(10), 38–45 (2011).
(36) T. Arakawa, D. Ejima, T. Li, and J.S. Philo, J. Pharm. Sci. 99(4), 1674–1692 (2010).
(37) D. Sheehan, R. FitzGerald, Protein Purification Protocols (Springer, New York, New York, 1996), pp. 145–150.
(38) D. Guillarme and M.W. Dong, Am. Pharm. Rev. 16(4), 36–43 (2013).
(39) T. Zhang, J. Bourret, and T. Cano, J. Chromatogr. A 1218(31), 5079–5086 (2011).
(40) J. Vlasak and R. Ionescu, Curr. Pharm. Biotechnol. 9(6), 468–481 (2008).
(41) C. Chumsae, K. Gifford, W. Lian, H. Liu, C.H. Radziejewski, and Z.S. Zhou, Anal. Chem. 85(23), 11401–11409 (2013).
(42) K.G. Moorhouse, W. Nashabeh, J. Deveney, N.S. Bjork, M.G. Mulkerrin, and T. Ryskamp, J. Pharm. Biomed. Anal. 16(4), 593–603 (1997).
(43) A.L. Pace, R.L. Wong, Y.T. Zhang, Y.-H. Kao, and Y.J. Wang, J. Pharm. Sci. 102(6), 1712-1723 (2013).
(44) Y.T. Zhang, J. Hu, A.L. Pace, R. Wong, Y.J. Wang, and Y.-H. Kao, J. Chromatogr. B 965(1218), 5079–5086 (2014).
(45) M.W. Dong, in Tryptic Mapping by Reversed Phase Liquid Chromatography, J.C. Giddings, E. Grushka, and R.P. Brown, Eds. (Marcel Dekker. New York, New York, 1992), pp. 21–51.
(46) S. Fekete, D. Guillarme, and M.W. Dong, LCGC North Am. 32(6), 420–433 (2014).
(47) S. Fekete, S. Rudaz, J.-L. Veuthey, and D. Guillarme, J. Sep. Sci. 35(22), 3113–3123 (2012).
(48) T.M. Dillon, P.V. Bondarenko, D.S. Rehder, G.D. Pipes, G.R. Kleemann, and M.S. Ricci, J. Chromatogr. A 1120(1), 112–120 (2006).
(49) T.M. Dillon, M.S. Ricci, C. Vezina, G.C. Flynn, Y.D. Liu, D.S. Rehder, M. Plant, B. Henkle, Y. Li, S. Deechongkit, B. Varnum, J. Wypych, A. Balland, and P.V. Bondarenko, J. Biol. Chem. 283(23), 16206–16215 (2008).
(50) D.S. Rehder, T.M. Dillon, G.D. Pipes, and P.V. Bondarenko, J. Chromatogr. A 1102(1), 164–175 (2006).
(51) T. Zhang, J. Zhang, D. Hewitt, B. Tran, X. Gao, Z.J. Qiu, M. Tejada, H. Gazzano-Santoro, and Y.-H. Kao, Anal. Chem. 84(16), 7112–7123 (2012).
(52) M. Zachariou, Affinity Chromatography: Methods and Protocols (Springer, New York, New York, 2007).
(53) C. Loew, C. Knoblich, J. Fichtl, N. Alt, K. Diepold, P. Bulau, P. Goldbach, M. Adler, H.-C. Mahler, and U. Grauschopf, J. Pharm, Sciences 101(11), 4248–4257 (2012).
(54) T. Schlothauer, P. Rueger, J.O. Stracke, H. Hertenberger, F. Fingas, L. Kling, T. Emrich, G. Drabner, S. Seeber, J. Auer, S. Koch, and A. Papadimitriou, mAbs 5(4), 576–586 (2013).
(55) C. Quan, E. Alcala, I. Petkovska, D. Matthews, E. Canova-Davis, R. Taticek, and S. Ma, Anal. Biochem. 373(2), 179–191 (2008).
(56) K. Eriksson and M. Belew, Hydrophobic Interaction Chromatography (John Wiley & Sons. Hoboken, New Jersey, 1998), pp. 283–309.
(57) A. Wakankar, Y. Chen, Y. Gokarn, and F.S. Jacobson, MAbs 3(2), 161–172 (2011).
(58) J. Cacia, R. Keck, L.G. Presta, and J. Frenz, iochemistry 35(6), 1897–1903 (1996).
(59) A.A. Wakankar, R.T. Borchardt, C. Eigenbrot, S. Shia, Y.J. Wang, S.J. Shire, and J.L. Liu, Biochemistry 46(6), 1534–1544 (2007).
(60) L.W. Dick, D. Qiu, R.B. Wong, and K.-C. Cheng, Biotechnol. and Bioeng. 105(3), 515–523 (2010).
(61) J. Valliere-Douglass, L. Jones, D. Shpektor, P. Kodama, A. Wallace, A. Balland, R. Bailey, and Y. Zhang, Anal. Chem. 80(9). 3168–3174 (2008).
(62) J. Valliere-Douglass, A. Wallace, and A. Balland, J. Chromatogr. A 1214(1–2), 81–89 (2008).
(63) Y. Yang and X. Geng, J. Chromatogr. A 1218(49), 8813–8825 (2011).
(64) J. Yang, S. Wang, J. Liu, and A. Raghani, J. Chromatogr. A 1156(1–2), 174–182 (2007).
(65) C. Wong, C. Strachan-Mills, and S. Burman, J. Chromatogr. A 1270, 153–161 (2012).
(66) T. Tetaz, S. Detzner, A. Friedlein, B. Molitor, and J.-L. Mary, J. Chromatogr. A 1218(35), 5892–5896 (2011).
(67) A.J. Alpert, J. Chromatogr. A 499, 177–196 (1990).
(68) J. Ahn, J. Bones, Y.Q. Yu, P.M. Rudd, and M. Gilar, J. Chromatogr. B 878(3–4), 403–408 (2010).
(69) A. Zhu, J. Martosella, and P.T. Duong, The Application Notebook, supplement to LCGC North Am. 31(s6), 27–29 (2013).
(70) B. Wei, B.J. Rogers, and M.J. Wirth, J. Am. Chem. Soc. 134(26), 10780–10782 (2012).
(71) M. Gilar, A.E. Daly, M. Kele, U.D. Neue, and J.C. Gebler, J. Chromatogr. A 1061(2), 183–192 (2004).
Taylor Yonghua Zhang is a senior scientist and a Group Leader in Protein Analytical Chemistry at Genentech in South San Francisco, where he has worked since 2006. He was formerly a research specialist and senior analytical biochemist at Dow Chemical. He received his PhD in Analytical Chemistry from Iowa State University in 2000. He has more than 10 years of experience in bioprocess development and has authored 20+ publications in the field of bioanalytical chemistry. Dr. Zhang's research interests involve utilizing different separation techniques for therapeutic protein characterization and developing methods for the quality control system. He is also the critical quality attribute (CQA) expert team lead for Genentech/Roche quality by design (QbD) efforts.
Cynthia Quan is a scientist in the Protein Analytical Chemistry Department at Genentech, a member of the Roche Group, located in South San Francisco. She is responsible for analytical characterization, method development, and technical transfers for clinical and commercial products, and for evaluation and implementation of new analytical techniques into legacy commercial control systems. She has more than 20 years of experience in bioprocess development, and has authored seven Chemistry, Manufacturing and Control (CMC) sections for clinical and commercial product regulatory filings, including Investigational New Drug (IND) and post-approval supplements. She has authored or coauthored 14 publications in the bioanalytical chemistry field.
Michael W. Dong is a senior scientist in Small Molecule Drug Discovery at Genentech in South San Francisco, California. He is responsible for new technologies, automation, and supporting late-stage research projects in small-molecule analytical chemistry and QC of small-molecule pharmaceutical sciences. He holds a PhD in analytical chemistry from the City University of New York and a certificate in Biotechnology from U.C. Santa Cruz. He has conducted numerous courses on HPLC/UHPLC, pharmaceutical analysis, HPLC method development, drug development process, and drug quality fundamentals. He is the author of 100+ journal articles and Modern HPLC for Practicing Scientists as well as a co-editor of Handbook of Pharmaceutical Analysis by HPLC. He is a member of the editorial advisory board of LCGC North America.


Michael W. Dong






 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")
, machine learning and modern computer technologies concepts. Business, Technology, Internet and network concept. | Image Credit: © photon_photo - stock.adobe.com")


A Well-Written Analytical Procedure for Regulated HPLC Testing
October 15th 2024This paper describes the content of a well-written analytical procedure for regulated high-performance liquid chromatography (HPLC) testing. A stability-indicating HPLC assay for a drug product illustrates the required components for regulatory compliance, including additional parameters to expedite a laboratory analyst’s execution.

ISC 2024: An Interview with Amarande Murisier
October 8th 2024As part of our ISC 2024 coverage, we recently interviewed Amarande Murisier of the University Hospital of Lausanne, Switzerland (CHUV) about her winning the Rising Stars of Separation Science Award for Biopharmaceutcal Analysis and her scientific background.


