How Much Can I Inject? Part I: Injecting in Mobile Phase
LCGC North America
What symptoms might indicate we have an injection problem?
Both the volume of the sample injection and the solvent in which the sample is dissolved can affect the appearance of the chromatogram.
At first glance, injection of sample onto a liquid chromatography (LC) column seems pretty simple — just put the sample in a vial, select the desired injection volume in the autosampler settings, and push the start button. However, before starting an LC method, we should check a few variables to be sure that we are not creating a potential problem. How does the injection volume affect the chromatogram? What about the injection solvent? Does the column size or packing particle diameter make any difference? What symptoms should we watch for that might indicate we have an injection problem? In this month's "LC Troubleshooting" installment, we'll look at a few of these factors so that we can understand what is going on and how to avoid problems.
It Really Is Quite Simple
Let's start with a conceptual look at the effect of injection volume on the appearance of a peak in the chromatogram. Throughout this discussion I'll assume ideal, Gaussian shaped peaks, and apply distortion to them. I will also assume that the sample concentration is sufficiently small that neither the column or the detector experiences overload. The results will vary a bit with real peaks, which tend to tail somewhat, and are influenced by some LC system characteristics external to the column. Finally, in the present discussion, I will consider only isocratic separations, where the mobile-phase composition is constant; next month we'll look at what happens in a gradient.


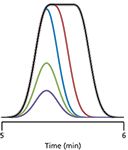
Figure 1: Simulated chromatograms showing the effect of increased injection volume on peak shape. Purple, green, and blue peaks for injection volume ratios of 1, 2, and 4, respectively, when no injection-related peak broadening is present. Red and black peaks represent mild and severe peak broadening because of excessive injection volume. Arbitrary time scale. See the text for details.
If we start with a small injection volume of sample dissolved in the mobile phase, we might expect a peak such as the purple one in Figure 1. As we double the injection volume, then double it again, we would expect the peak to grow in height and area, as with the green and blue peaks, respectively. As the peak grows under nonoverload conditions, the column plate number, N, will stay constant. N is defined as


where tR is the retention time and w is the baseline width of the peak between tangents drawn to the sides of the peak. If the peak is Gaussian in shape, the present assumption, the width will be 4 standard deviations, 4σ. If the plate number stays constant as the peak grows, and the retention time does not change, this means that the peak width will also stay constant. This can be seen as the peaks of Figure 1 grow from the purple to green to blue tracings.
With a sufficiently small injection volume, we can assume that all the sample molecules arrive at the head of the column at the same time, and as they migrate down the column, the peak spreads into the familiar Gaussian shape. If the peak is too large, on the other hand, the sample molecules on the leading edge of the peak arrive at the column before those at the trailing edge. Because we're assuming that the injection solvent is identical to the mobile-phase composition, we can imagine that those first-on molecules get a head start in the chromatographic process and move down the column a bit before the last-on molecules get started. The sample molecules on the leading edge of the large injection behave exactly as they would in a smaller injection, so the front edge of the peak appears unchanged. You can see this in Figure 1, where the leading (left-hand) edge of the blue, red, and black peaks overlap perfectly. With a large injection, once the peak height reaches its maximum, the sample concentration is constant, and will remain so until the tail of the injection band is reached. Thus, when too large a sample volume is injected, the peak rises normally, then is flat-topped, then drops back to the baseline with the normal Gaussian tail. I have shown two different large-volume injections in Figure 1, red and black, where the tail (right edge) looks identical to the blue one, but offset to a later time. You can think of this as inserting a constant-concentration band in the middle of the peak, pushing the back edge to a later retention time. For the red peak, the injection volume is just a bit too large and the peak broadens and appears rounded on the top, but the black peak is quite a bit too large and the flat top on the peak is obvious. You can imagine that if the injection volume is big enough, the molecules on the leading edge of the peak could reach the detector before those on the trailing edge reach the inlet of the column.
If we have two components in our sample, each one behaves independently. In Figure 2, I have shown what happens under the conditions of Figure 1 when two peaks are present. At small injection volumes, the peak sizes grow with injection volume for the purple, green, and blue peaks of Figure 2. Because there is no overload, the separation stays the same, so the valley between the peaks does not move. (The slight offset in the valley for the purple, green, and blue traces is an aberration in the way I smoothed the data.) In the present example, I have set the resolution (Rs) to 1.5, where the valley just touches the baseline (read more about resolution in last month's installment [1]). When the injection volume is too large, as we saw above, the front edge of the peak behaves normally, but the back edge is shifted to a later time; this happens for each component of the sample. So in Figure 2, for the red and black cases of too large an injection, the front edges of each peak look just as they did for the blue peak. The molecules at the back edge of the first peak of the red and black examples are still at a sufficiently large concentration when the second peak begins to be eluted so that the valley between the peaks is raised and shifted to a later time, thus reducing resolution. This loss in resolution is small, but noticeable for the red case, but for the black tracing, the injection volume is much too large, and the loss of resolution is severe. Note also that the apparent peak center (retention time) and valley shift to longer times as the magnitude of the excess injection volume increases.
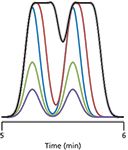
Figure 2: Simulated chromatograms showing the effect of increased injection volume on resolution. Same peak coding and conditions as in Figure 1. See the text for details.
So to recapitulate, as we increase the injection volume, the peak height and area grow in proportion to the injection volume up to the point when the injection volume is too large. At that point, the peak broadens, and this peak broadening can reduce resolution between two adjacent peaks. The questions now are: How much can we inject before we inject too much? How much loss in resolution can we tolerate?
And Now the Numbers
Above we saw conceptually what happens when the injection volume becomes too large. Now we need to find out what happens quantitatively. (If you don't care about the calculations, skip to the next section.) You'll recall that resolution (Rs) is calculated as:
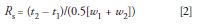
where t1 and t2 are the retention times of the two peaks, respectively, and w1 and w2 are the corresponding baseline peak widths. If the peaks are closely eluted, they will have approximately the same peak width, so we can simplify the denominator of equation 2 to the average peak width, w, where w ≈ w1 ≈ w2. For the present discussion, where we are interested in injection volume, it is more convenient to consider resolution in terms of volumes:
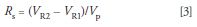
where VR1 and VR2 are the retention volumes of peak 1 and peak 2, respectively and Vp is the average peak volume (Vp = w × F, where F is the flow rate).
The peak volume observed in a chromatogram has two primary contributions, the volume (width) resulting from normal Gaussian band-broadening processes within the column, and the volume resulting from injecting too large a sample (as in the red and black samples of Figures 1 and 2):

where Vs is the volume of sample injected, and Vp0 is the volume of the peak for a very small volume sample (for example, the purple or green peaks of Figures 1 and 2). You can see that the resulting peak volume is simply the square root of the sum of squares of each contribution, with a little extra broadening for the injection volume because of factors (2) that we don't need to go into here.
We can determine Vp0 from the retention volume (time) of a peak, VR, and the plate number, N, of the column:

The retention volume is calculated from the retention factor, k, and the column dead volume, VM:

And the dead volume (in milliliters) can be estimated from the column length, L, and diameter, dc (both in millimeters):
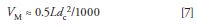
We then can combine equations 5, 6, and 7 to allow us to estimate Vp0 from the column dimensions, plate number, and sample k-value:
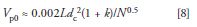
As has been discussed in "LC Troubleshooting" many times in the past, it is best to have k ≥ 1 to take maximum advantage of the column and to avoid interferences with the solvent front. For the present discussion, we'll assume k = 1; the effects generally will be more severe with k < 1 and less so with k > 1.
Now, we have all the tools we need to determine the effect of the injection volume on resolution using equations 3, 4, and 8. We can measure retention volumes, VR, from the chromatogram and estimate Vp0 with equation 8. The observed peak volume, Vp, can then be estimated for different sample volumes, Vs, with equation 4.
Some Examples
Next, let's look at the effect of injection volume on five popular column sizes, three commonly used for conventional LC (<6000 psi [400 bar]) with lengths of 150 or 100 mm, diameters of 4.6 or 2.1 mm, and packed with 5- or 3-µm diameter, dp, particles; one for LC with mass spectrometric detection (LC–MS), 50 mm × 2.1 mm with 3-µm particles; and one for ultrahigh-pressure LC (UHPLC, >6000 psi [400 bar]), a 50 mm × 2.1 mm column containing 1.7-µm particles. These are summarized in Table I. I'll refer to these by their identification numbers 1–5 in the left-hand column of Table I. I have included estimates of realistic plate numbers, column dead volumes, retention volumes (for k = 1), and peak volumes for small peaks, all determined from the equations referenced in the footnote to the table. (I'll issue my standard disclaimer here: The numbers in the various tables have been rounded or truncated for display purposes, so if you repeat these calculations, your results may vary slightly from mine.)

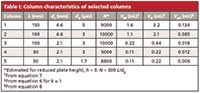
Table I: Column characteristics of selected columns
In Table II, I've shown the calculated percent loss in resolution for injection volumes of 1–1000 µL for the columns of Table I. We can see some general trends in the data. If we pick some arbitrary value for the acceptable loss in resolution, such as 5%, we can see that the most popular column for conventional LC, column 1, can tolerate between 20 and 50 µL of sample injected in mobile phase. Anything that is done to reduce the peak volume generated by the column will also reduce the acceptable injection volume. For example, column 2 has approximately the same plate number as column 1, so it should give the same separation, but its smaller volume will require a reduction in injection volume to something closer to 20 µL. If we try to conserve solvent and move to column 3, which is just a smaller diameter version of column 2, the column will tolerate only 5 µL of sample. For UHPLC, column 5 will require that we reduce the volume even further.


Table II: Percent resolution loss for various columns and injection volumes
Table II is useful if we want to know how much of an impact a given injection volume will have for a particular column configuration. An alternative way to look at the same data is shown in Table III, where I've shown the maximum volume of sample (in microliters) that can be injected in the mobile phase for a given loss in resolution (1%, 5%, or 10%) with each column configuration. For the example above, where we considered a 5% loss in resolution, column 1 can tolerate 37 µL of sample, whereas column 5 needs to be restricted to 2.6 µL.

Table III: Maximum injection volume for a specified loss in resolution
Although it is nice to have tables to use for data lookup, I find that it is even nicer to have a general rule of thumb that I can apply. If such rules are simple, they are easy to remember and apply even when the table isn't handy. We can create such rules by comparing the allowable sample volumes, Vs, in Table III with the small-injection peak volumes, Vp0, for the corresponding conditions in Table I, as summarized at the bottom of Table III. From this, we can see that for a 1% loss in resolution, Vs ≈ 0.15 Vp0. That is, if we inject using mobile phase as the injection solvent, we can inject up to ~15% of the volume of the first peak of interest, which I call the 15% Rule. Similarly, if we can tolerate a 5% loss in resolution, the factor is ~30% of Vp0, and for a 10% loss, ≈40% of Vp0. I like to work on the conservative side of things, and I've often used the 15% Rule in "LC Troubleshooting" when recommending injection volumes. By using these rules, you don't have to worry about the k-value or retention times — just look at the volume of the first peak of interest by converting a small injection peak width from time to volume (Vp0 = w × F).
Summary
In this discussion, we've looked in detail at what happens to the sample peak characteristics as we inject different volumes of sample dissolved in mobile phase onto an LC column under isocratic conditions. We've seen that the appearance of the resulting peaks is dependent on the peak volume (width) of a small-injection sample, which in turn is dependent on the length and diameter of the column, as well as the packing particle size and peak retention (k-value). As a general rule, any system change that reduces the volume of the peak generated by the column (shorter column, narrower column, smaller packing particles, or shorter retention) will also reduce the volume of sample we can inject before resolution is degraded. Based on the data presented here, we were able to generate some practical rules of thumb, such as the 15% Rule, which states that for a 1% loss in resolution, we can inject up to 15% of the volume of the first peak of interest if we inject in mobile phase.
The discussion assumed that we were using the mobile phase as the injection solvent. If a stronger (higher percent organic in reversed-phase LC) or weaker (higher percent aqueous) injection solvent is used, or the method uses gradient conditions, the results will vary. We'll consider these in next month's installment. We've also assumed that neither the column nor the detector was overloaded, either of which can change the appearance of the peaks. Finally, we've assumed that the injection volume is the only added factor contributing to peak broadening, which is rarely the case. From a practical standpoint, the injection band can broaden by as much as 50% (3) as it is washed from the sample injection valve. Additionally, extracolumn band broadening because of the system plumbing, detector cell design, and other factors can play a role in the observed peak widths.
When all the above factors are taken into play, the recommended injection volumes in Tables II and III, or the rules of thumb should be interpreted as guidelines for a good place to start. I recommend doing a simple empirical test to see if the selected conditions are acceptable. Pick a target injection volume for the maximum desired loss in resolution, such as from Table III, then inject this volume plus an injection twice as large and half as large. If the results are acceptable for all three injections, the target volume should be safe. If you see significant changes in resolution or peak shape between the injections, you may need to further evaluate if the injection volume is sufficiently robust for your purposes.
References
(1) J.W. Dolan, LCGC North Am. 32(9), 718–725 (2014).
(2) J.C. Sternberg, Adv. Chromatogr. 2, 205 (1966).
(3) L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography, 3rd ed. (Wiley, Hoboken, New Jersey, 2010), p. 70.
John W. Dolan "LC Troubleshooting" Editor John Dolan has been writing "LC Troubleshooting" for LCGC for more than 30 years. One of the industry's most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources in Walnut Creek, California. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com
John W. Dolan






: An Interview With Fabrice Gritti")




, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.


Using GC-MS to Measure Improvement Efforts to TNT-Contaminated Soil
April 29th 2025Researchers developing a plant microbial consortium that can repair in-situ high concentration TNT (1434 mg/kg) contaminated soil, as well as overcome the limitations of previous studies that only focused on simulated pollution, used untargeted metabolone gas chromatography-mass spectrometry (GC-MS) to measure their success.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

Potential Obstacles in Chromatographic Analyses Distinguishing Marijuana from Hemp
April 28th 2025LCGC International's April series for National Cannabis Awareness Month concludes with a discussion with Walter B. Wilson from the National Institute of Standard and Technology’s (NIST’s) Chemical Sciences Division regarding recent research his team conducted investigating chromatographic interferences that can potentially inflate the levels of Δ9-THC in Cannabis sativa plant samples, and possible solutions to avoid this problem.

