How to Reduce Mobile-Phase Consumption
LCGC North America


Are your solvent costs too high? Here are some ideas about how to decrease the amount of mobile phase you use.
Are your solvent costs too high? Here are some ideas about how to decrease the amount of mobile phase you use.
In today’s laboratories, expense reduction has become a standard part of doing business. In some cases, the amount of money we spend on solvents for the mobile phase in liquid chromatography (LC) operations can be a significant part of the cost of analysis. In other cases, it can be less of a concern. For this month’s “LC Troubleshooting” discussion, I’d like to look at some of the options that may be appropriate ways to reduce the cost of mobile-phase solvents in your laboratory. There are three major ways to approach the problem: we can recycle all or part of the mobile phase and use it again, we can recover solvents through distillation of the waste mobile phase, and we can reduce the amount of mobile phase that we send through the column. I’ll consider each of these techniques below. Sometimes a combination of techniques may be used for further gains in cost reduction. Be aware that not all of these techniques may be available for your application. For example, direct recycling will work only for isocratic separations and not gradients.
Recycle
In my community, we have at-curb recycling for part of our household waste. This reduces the amount of waste piling up in the landfill and recovers certain materials, such as aluminum, steel, paper, and some plastics, for reuse. We are also encouraged to reduce the amount of waste we generate by reusing some materials and using less of others. When I return beverage containers to the grocery store, my deposit on these is returned. In some cases, the whole recycling process reduces my expenses, whereas in others it is done because it is the right thing to do from an environmental stewardship standpoint.
When we consider the mobile phase used in LC operations, it may be possible to recycle all or some of the components, but even if it is possible, the economics of the process may not make practical sense. If your method is isocratic, the mobile-phase composition is constant, and you can directly recycle your mobile phase in many cases. This does not work for gradient methods, because the waste bottle will contain an “average” mixture of the mobile phase that is not directly useful.
The simplest way to recycle the mobile phase is to direct the waste line from the detector directly back into the reservoir. This seems like a rash decision and many workers are reluctant to do this, even though it can be done successfully for many methods. “But,” you ask, “aren’t you contaminating the mobile phase with sample components that are eluted from the column?” Indeed you are, but let’s consider what is happening. Under isocratic conditions, the mobile-phase composition is constant, and even if you have a retained component of the mobile phase, it will not generate a peak in the chromatogram. For example, most ion-pairing reagents are chosen because they stick strongly to the column and build up a charge on the column packing. Under the steady state conditions of isocratic flow, you don’t see a peak for the ion-pairing reagent in the chromatogram. However, if you were to inject a sample that contained that same ion-pairing reagent-for example, hexane sulfonate-into a mobile phase that did not contain that compound, you might see a peak for the reagent if the detector were set to detect it. In other words, if a compound is introduced to the column at a steady state, its detector response will be constant and no peaks will be observed. This is exactly what happens if you direct the waste stream back into the reservoir.
There are a few things to be aware of to minimize potential problems with direct recycling of mobile phase. First, you would like the mobile-phase composition to be constant and homogeneous. If you are continually adding sample waste components, the composition will not be truly constant, but if the dilution factor is sufficient, it will be constant from a practical standpoint. For example, if you have 1 L of mobile phase and you add 1 mL/min of waste to it while pumping 1 mL/min out of the reservoir, the volume will stay the same, but the composition will change very slowly over time. In this example, the already-diluted sample that is eluted from the column would be diluted 1000-fold more as it entered the reservoir. The background signal will gradually increase as more and more analyte is added to the reservoir, but this generally will not be noticeable over a few days’ time. To keep the solution as homogeneous as possible, place the reservoir on a stir plate and add a stir bar to keep it well mixed. Just as you need to be careful not to use a single preparation of mobile phase for too long without replacement so as to avoid evaporation, microbial growth, or other problems, you can’t use recycled mobile phase forever, either. You’ll have to determine how long you can use the mobile phase, but I’d suggest a maximum of 1–2 weeks. Generally, I prefer to make fresh buffers at least once a week even without recycling, and recycling will certainly not extend your current expiration policies.
Direct recycling generally will be most successful when your analytes generate a large signal-to-noise ratio. It is most likely that any problems related to recycling will appear first for trace levels of analytes, such as with an impurity method, so direct recycling may not be practical for these methods.
Because direct recycling gradually contaminates the mobile phase, an alternate recycling technique can be used, which I’ll call fractional recycling. If you consider a normal day’s LC operation in the laboratory, you put the mobile phase on the system and allow the column to equilibrate. Then you run a system suitability test, followed by some calibration standards, and finally run your samples for analysis, perhaps with occasional standards or check samples added. Of the total time of operation, for only a small fraction of the time is there a sample component being eluted from the column. If the fractions of the mobile phase that contain analytes and unwanted peaks, as well as the material eluted at the column void (solvent front), could be directed to waste, the remaining mobile-phase stream should be pure. If you put a two-way valve after the detector, you could watch the chromatogram, and when peaks were being eluted, you could direct the waste stream to a waste container; when no peaks were being eluted, you could direct the waste stream back to the sample reservoir. If you do this manually, of course, the labor costs will quickly eclipse any savings realized by reducing solvent consumption. What you need instead is a device that looks at the detector output and determines if a peak is present or not-this is what your data system does. By using a similar device to detect peaks and control the switching valve, you could have an automated recycler to replace the person running a manually controlled valve. Such a device is available from Spectrum Chromatography, called the S-3 HPLC Solvent Recycler. (I’m sure similar devices are available from other sources, as well.) The manufacturer claims that if you are able to recycle 25 L of high performance liquid chromatography (HPLC)-grade acetonitrile, you will pay for the device. If you use it, I’d suggest taking the same precautions I mentioned above for direct recycling: Use a stir bar to keep the reservoir contents homogeneous, and don’t try to extend the expiration date of your mobile phase beyond current limits.
As an alternate technique, you could use an automated switching valve and control it with the timed events output from your LC system. If you diverted the timed section of the chromatogram at the solvent front and perhaps where large peaks eluted, you would get the major advantages of fractional recycling without having to buy a specialized instrument. However, the commercial recyclers don’t cost much more than an automated switching valve, so it may not be worth your time to design your own device.
Distill
When I started doing LC analyses in a pesticide residue laboratory in graduate school, our department distilled all our solvents to increase their purity. Today most of us simply purchase high-purity, HPLC-grade solvents. However, you could use distillation to recover the solvents from the waste mobile phase, perhaps with a spinning-band distillation column to get added purity. This would allow you to recover the organic solvent from an aqueous–organic mobile-phase waste stream that also contained buffers or other additives. Because distillation separates the organic solvent for recovery, it will work just as well for isocratic and gradient LC waste streams.
At least one company, B/R Instrument Corp., offers several different distillation products to allow you to recover mobile-phase solvents. These recovered solvents then can be used to prepare fresh mobile phases. If I were to use one of these devices, I would like to be confident that it produces sufficiently pure solvents for LC analysis, especially if you are performing trace analysis with gradient elution. I’m sure that the noted supplier (or others) would be happy to discuss this concern with you.
Reduce
Perhaps the easiest way to conserve solvent is to reduce how much you use. You can make most of the changes discussed below and remain within the guidelines that the pharmacopeias recommend. This means that all you need to do is verify that the new conditions give the same results as the original, document these results, and proceed with analysis. Your company may require additional procedures or documentation when such changes are made.
As a general rule, if you keep the column chemistry, column plate number, and mobile-phase composition constant, you should obtain the same separation. Special attention needs to be paid to gradient methods, which may require compensating changes in the conditions, and for cases when instrument extra-column volume plays a significant role in band width. Next let’s look at three possible approaches to reduce mobile-phase use.
Column Diameter
Perhaps the simplest way to reduce solvent consumption is to reduce the column diameter. If the flow rate is adjusted for the change in column cross-sectional area, you should get the same separation and retention times with both isocratic and gradient methods. Take, for example, a method running on the most popular column, a 150 mm × 4.6 mm column, operated at a flow rate of 2.0 mL/min. We generally can reduce the column diameter to other popular sizes of 3.0 or 2.1 mm i.d. without excessive extracolumn band broadening. For the 3.0-mm column, the reduction in cross-sectional area is (3.0/4.6)2 = 0.4. To keep the same linear velocity of the mobile phase through the column, we need to reduce the flow rate by this same factor, so (2.0 mL/min × 0.4) = 0.8 mL/min. (As usual for these discussions, I’ve rounded or truncated calculated values, so your results may vary slightly if you repeat my calculations. Also, to avoid clutter I’ve not included units in some of the examples where they should be obvious.) This should give the same retention times as the original conditions, but with approximately 60% reduction in solvent used. In a similar manner we can calculate the effect of changing to a 2.1-mm i.d. column: (2.1/4.6)2 = 0.2. Apply this to the flow rate, (2.0 mL/min × 0.2) = 0.4 mL/min, and you’ll reduce your solvent consumption by 80%. You could use even smaller diameter columns, but if you reduce the diameter below 2.1 mm, it is likely that you’ll have excessive band broadening because of extracolumn effects unless you modify the LC instrument.
Column Length and Particle Size
A reduction in column packing particle diameter (dp) will increase the column plate number inversely to the change in particle diameter. This, then, will allow you to use a shorter column for the same plate number. Take the example above for a 150-mm-long column packed with 5-µm dp particles. If we change to 3-µm particles, this will increase the plate number by 5/3 = 1.67-fold. For a constant plate number, we can reduce the column length by the same factor: (150 mm/1.67) = 90 mm. Because 90-mm columns aren’t commonly available, we’d round this length to 100 mm. This tells us that the 100-mm column packed with 3-µm dp particles would have approximately the same plate number as the 150-mm, 5-µm column. But because the length has been reduced by one-third, isocratic retention times would be reduced by one-third also, as would the method run time, so we’d save one-third of the solvent costs. In this example, the smaller particles will increase the pressure by (5/3)2 = 2.8-fold and the shorter length will reduce the pressure by 100/150 = 0.67-fold, resulting in an overall pressure increase of (2.8 × 0.67) = 1.8-fold. Many methods operate at pressures where this increase would still be within the instrument’s pressure capabilities. You could lower the flow rate to reduce the pressure; this would increase the retention times, but would not change the amount of solvent used per run. For example, reducing the flow rate 1.8-fold would mean operating at (2.0 mL/min /1.8) = 1.1 mL/min. A flow rate of 1.1 mL/min with a 100 mm column would give retention times of (0.67 × 1.8) = 1.2-fold larger than the original method, or a 20% increase in run time. An alternative would be to run at 1.5 mL/min for the same retention times and run time, but a 30% increase in pressure, which perhaps would be more acceptable. As discussed below, flow-rate changes beyond those used to generate constant linear velocity are appropriate for isocratic methods, but not for gradient methods without additional method adjustments.
Column Length and Diameter Plus Particle Size
By combining a change in column diameter with a change in the length and particle size, we can further reduce solvent consumption. Let’s consider that the original isocratic method used a 150 mm × 4.6 mm, 5-µm dp column operated at 2 mL/min and a run time of 10 min. This would use (2 mL/min × 10 min) = 20 mL of solvent per run. Reducing the column diameter to 2.1 mm was compensated for by reducing the flow rate to 0.4 mL/min for a solvent volume of (0.4 mL/min × 10 min) = 4 mL of solvent per run. If we combine this diameter change with the length and particle size change to a 100-mm, 3-µm column, we saw that this would reduce run times by 0.67. This would also reduce the total solvent used per run to (4 mL × 0.67) = 2.7 mL/run. We now would be using (2.7/20) ≈ 15% of the original solvent. I’ll leave you to calculate the change in pressure for this new condition.
The above changes in length, diameter, particle size, and flow rate will not change the separation for isocratic methods, but more care needs to be taken for gradient methods. For gradients, it is necessary to keep the number of column volumes of solvent constant for each gradient segment when the conditions are adjusted, using the following equation:


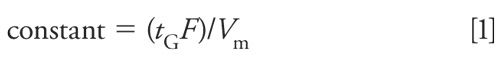
where tG is the time for each gradient segment (in minutes), F is the flow rate (in milliliters per minute), and Vm is the column volume (in milliliters). A reduction in Vm is proportional to the change in column length times the cross-sectional area (or diameter squared). This means that for two conditions (with subscripts 1 = original and 2 = new), we need to have

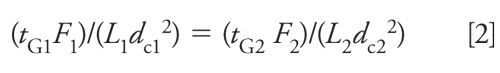
For any given set of changes, as proposed above, we can determine the necessary gradient time adjustment by solving equation 2 for the new gradient time, tG2:

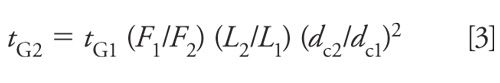
Let’s apply equation 3 to the above example, where we changed from a 150 mm × 4.6 mm, 5-µm dp column operated at 2.0 mL/min to a 100 mm × 2.1 mm, 3-µm dp column operated at 0.4 mL/min. Let’s further assume that the original gradient time, tG1, was 10 min. What is the new gradient time, tG2, that is required to get the same separation under the new conditions? Using equation 3, we get our answer: tG2 = 10 min (2.0/0.4) (100/150) (2.1/4.6)2 = 6.9 min
The original method used (2.0 mL/min × 10 min) = 20 mL of solvent; the new conditions use (0.4 mL/min × 6.9 min) = 2.8 mL. Thus, the revised method would use (2.8/20) ≈ 15% of the original solvent, the same savings as for the isocratic method. Note that unless we change the gradient time as in equation 3, the separation (peak spacing) will change. For a multistep gradient, the time for each gradient step will need to be adjusted using equation 3. Additional method tweaking may be required when peaks are eluted early in the gradient, because the system dwell volume can be important for peak spacing of early peaks.
The examples in this section assumed conventional LC conditions, but the same changes will apply equally for ultrahigh-pressure liquid chromatography (UHPLC) or changes from LC to UHPLC. Also, be aware that a smaller-volume column may require a reduction injection volume or injected sample mass to avoid volume- or mass-overload of the column.
Conclusions
Most of us are driven by the economic factors in the laboratory, so be sure to look at all of the costs when applying any of the solvent-saving techniques discussed above. For example, if you have to buy a new piece of hardware for $1000 or more, how many liters of solvent need to be saved to break even (and don’t forget the cost of labor)?
Usually the most practical and inexpensive way to reduce solvent consumption is by changing the column length, diameter, or particle size, as well as the flow rate. If you work in a regulated environment, such as pharmaceutical or environmental analysis, be sure that any changes are within the limits set by your company’s standard operating procedures (SOPs) and the regulatory guidelines. Documentation of such changes is essential for all applications; validation requirements will vary depending on the SOPs, the application, and the magnitude of the change involved.


John W. Dolan“LC Troubleshooting” Editor John Dolan has been writing “LC Troubleshooting” for LCGC for more than 30 years. One of the industry’s most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources in Lafayette, California. He is also a member of LCGC’s editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com










Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.

Mobile Phase Buffers in Liquid Chromatography: A Review of Essential Ideas
December 11th 2024In this installment of "LC Troubleshooting," Dwight Stoll discusses several essential principles related to when and why buffers are important, as well as practical factors, such as commonly used buffering agents, that are recommended for use with different types of detectors.


In-Loop Analyte Degradation in Two-Dimensional Liquid Chromatography: Example and Solutions
September 13th 2024In this installment of “LC Troubleshooting,” we describe an artifact that can arise because of compound degradation during the transfer of fractions of the first dimension (1D) column effluent to the 2D separation.


