Forensic Drugs Analysis: A Review of Miniaturized Separation Techniques
LCGC North America
Miniaturized separation techniques are advantageous for the analysis of illicit drugs and new psychoactive substances.
Miniaturization is one of the emerging trends in modern analytical chemistry. Capillary electrophoresis (CE), capillary electrochromatography (CEC), and more recently nano-liquid chromatography (nano-LC) have been successfully miniaturized and applied to forensic drug analysis. This review will discuss the recent applications of miniaturized separation techniques for the analysis of illicit drugs and new psychoactive substances in seized materials and biological fluids. Particular attention is given to chiral discrimination of abused drugs, which is an important topic in forensic analysis.
The abuse of addictive drugs is a complex problem globally, and involves political, cultural, and socioeconomic factors (1–3). A study estimated that the cost of illicit drug use in the United States in 2007 was more than $193 billion. This amount originated from direct and indirect costs in three main areas: crime, health, and productivity (4). From an historical point of view, the use of illicit drugs began after the second world war, particularly during the social upheaval of the 1960s, when cannabis became a symbol of the hippie revolution. Cocaine and crack abuse reached a peak in the 1980s, and in the 1990s there was an increase in recreational drug consumption amongst teenagers, particularly for ecstasy (5–7).


(PHOTO CREDIT: LAURENT HAMELS/GETTY IMAGES)
During the 1980s, government agencies began to strictly regulate and prohibit the use of addictive drugs (8–10). At the same time, new drugs with potential to be abused entered the illegal market. These new drugs included synthetic drugs universally known as “smart drugs,” and included synthetic chemicals related to cathinone, an amphetamine-like stimulant found naturally in the khat plant, and sold as “bath salts,” to enter the recreational drug market (11).
The identification and determination of illicit drugs in pills, as well as in biological fluids, has become a challenging task for analytical chemists.
In forensic analysis, two complementary analytical approaches have been adopted to monitor drugs of abuse. The first is based on a preliminary immunoassay screening, which has the advantage of speed, availability in on-site analysis kits, and high throughput. However, disadvantages include cross-reactivity, limited specificity, and false positive responses. For these reasons, confirmatory tests follow, generally performed by high performance liquid chromatography (HPLC) and gas chromatography (GC), coupled with mass spectrometry (MS). These methods also allow quantification of targeted compounds (12,13).
Recently, capillary electrophoresis (CE) and related techniques have demonstrated their potential in forensic analysis, offering high separation efficiency, the possibility of automation, reduced consumption of reagents, and low sample volume for analysis (14,15). CE is now a valid and accepted alternative, complementary to conventional chromatography. Furthermore, electrophoretic techniques can be matched with microfluidic devices for the on-site separation and qualitative–quantitative analysis of compounds of interest. This portability aspect would be very useful for law enforcement agencies to test for drugs of abuse in the workplace, or to test drivers suspected of being under the influence of illegal substances, or athletes during a sporting event (16).
The advantages of reduced costs for solvent and waste disposal have established the value of miniaturization in chromatography, where microcapillary, and nano-liquid chromatographic systems have gradually emerged (17). The number of applications of these chromatographic techniques is still very low in forensic analysis, but the results are promising for the future.
It is worth highlighting that miniaturized separation techniques, both electrophoretic and chromatographic, play a fundamental role in chiral separations. In this context, chiral discrimination of a drug or its metabolites (or both) can be useful for pharmacodynamic studies, evaluating toxic effects, and differentiating between the consumption of illegal (racemic) drugs or legal medicines containing only a single enantiomer (18).
The aim of this review is to report applications of miniaturized separation techniques from 2009 to the present in forensic analysis, strictly related to illicit drugs of abuse. Research present in the literature will be reported and will be regrouped according to major classes of abused drugs.


Analysis of Opioids, Opiates, and other Alkaloids
The United Nations Office on Drugs and Crime (UNDOC) reported that the global abuse of opiates remains essentially stable and continues to be one of the main problem drugs worldwide. Among opiates and their derivatives, heroin is still the most consumed narcotic drug. For this reason, the determination of heroin, its main metabolites, and its impurities represents a challenging task for analysts involved in forensic analysis. For many years, and mainly since the pioneering work of Lurie and colleagues (19), CE has become a widely accepted technique in this area.
The determination of the ratio of basic impurities of heroin such as morphine, codeine, O-3-monoacetylmorphine, O-6-monoacetylmorphine, acetylcodeine, noscapine, papaverine, and diacetylmorphine gives precious analytical information about the geographical origin of an illicit heroin sample. This topic was the objective of many CE studies, where the use of coated capillaries was mandatory for the analysis of such basic compounds (20). Recently, the use of stable and water-soluble gold nanoparticles was proposed as a stable coating for capillaries. This allowed coupling with the MS detector and the development of a CE-MS method for the analysis of heroin and its related alkaloids, suitable for the identification of geographical origin of illicit samples (21).
In recent years, the abuse of prescription opioids, such as fentanyl, or of substitution opioids such as methadone, has become a serious problem. As a result many clandestine laboratories started to produce fentanyl derivatives, even those not always approved for use in human beings. A non-aqueous CE–MS2 (NACE–MS2) method was proposed for the identification of fentanyl derivatives, which were detected down to the nanomolar level (0.5 ng/mL for fentanyl). The method was successfully applied to the analysis of three samples from forensic casework (22).
As proof of the fast analysis achievable with electrophoretic techniques, a rapid screening of 12 narcotic drugs was obtained using a microfluidic device with micellar electrokinetic chromatography (MEKC) in 200 s. This method resulted in a promising alternative for forensic analysis, with the advantages of low cost, a reduction in the consumption of reagents and samples, and a short separation time. The applicability of the method was further demonstrated by analyzing urine samples spiked with the studied drugs, and subjected to a liquid–liquid extraction (LLE) (23).
The main problems in determining drugs of abuse and their metabolites in biological fluids lies in interfering endogenous compounds, the time required by the sample preparation steps, and manipulation of low sample volumes. To solve these issues, many strategies were adopted for miniaturized techniques that are appropriate for the handling of a small volume of samples. The aim of these strategies is to guarantee appropriate limits of detection for miniaturized techniques, and ensure that they are comparable with those offered by conventional tools.
An in-line solid-phase extraction (SPE)–CE method was developed for the determination of 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP), codeine, hydrocodeine, and 6-acetylmorphine in water and in urine. UV detection limits for the first application were between 50 µg/mL and 200 µg/mL (24). CE was coupled with MS to optimize the parameters affecting the separation and the hyphenation, and reached detection limits in the 0.013–0.210 ng/mL range in the case of urine analysis, with a gain in sensitivity of about 100–200-fold (25).
Another study demonstrated the possibility of obtaining limits of detection (LODs) at the picogram-per-milliliter level for the same compounds, by combining in-line extraction with MS detection, and the use of an injected sample volume of 30 times the capillary volume (26).
As an alternative to preconcentration systems based on chromatographic mechanisms, electrophoretic can be exploited, particularly in CE. A pH-mediated stacking technique was used for the analysis of opiates in human saliva (27). The method provided 1000-fold sensitivity enhancement compared with the normal hydrodynamic injection, resulting in detection limits lower than 10 ng/mL.
Another widely used illicit drug is cannabis. Δ9-Tetrahydrocannabinol (THC) is the primary psychoactive component in cannabis and is mainly metabolized to 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid (THC-COOH), which is used in the forensic field to prove the use of cannabis. Alternatively, THC-COOH glucuronide (THC-COOH-glu), a phase II metabolite, was proposed because it can supply additional information such as the THC-COOH-glu/THC-COOH ratio in urine, which is useful to assess the frequency of cannabis use. The simultaneous detection of THC-COOH and THC-COOH-glu in urine samples collected from cannabis users has been performed with CE–MS (28). The validated method allowed LODs of 50 ng/mL to be achieved, and the only pretreatment needed for the urine sample was dilution with methanol and centrifugation.
The recent trend related to consumption of biogenic drugs, which mostly originate from traditional medicines and are often sold as herbal blends on the internet, has opened up a new avenue for forensic investigation. Strychnine and brucine alkaloids can be used as adulterants of street drugs, and cases of intoxication or even death have been reported. Field-amplified sample stacking (FASS) has been applied for their simultaneous determination in human urine by CE, and a detection limit as low as 2.5 ng/mL has been achieved (29).
Mesembrine alkaloids from Sceletium tortuosum, a plant endemic to South Africa and from which the drug Kanna is obtained, were determined by NACE–MS and the analysis of samples from vendors, self-fermented samples, and products ready for consumption produced a good overview of the chemotypic differences of plant samples as well as Kanna preparations (30).
As well as their use as recreational drugs, many compounds, including some alkaloids, are used in drug-assisted robberies and drug-assisted sexual assaults. This is the case for scopolamine, a tropane alkaloid, which is flavorless and odorless and can be delivered orally, dermally, or via inhalation. A portable device for CE with contactless conductivity detection was developed for the rapid determination of scopolamine and its separation from atropine in samples, such as an infusion of Datura stramonium L., spiked moisturizing cream, and spiked alcoholic beverages. The method was supported by the development of simple and fast sample pretreatments (31).
Analysis of Cocaine and its Metabolites
Cocaine, the major alkaloid of Erythroxylum coca, is one of the most widely consumed drugs of abuse in the world. Cocaine is a potent central nervous system stimulant that causes feelings of euphoria, increased self-esteem, and a state of alert. However, chronic abuse leads to side effects such as states of depression, hallucinations, cardiac arrhythmias, respiratory problems, and toxicity of target organs such as liver and kidney (32–35).
The monitoring of cocaine abuse is important in both toxicological and forensic analysis. In recent years, cocaine detection has been investigated in cases such as driving under the influence of drugs, criminal justice, and workplace drug-testing programs.
The major routes of illicit cocaine administration are intranasal, intravenous, and smoking. Cocaine is metabolized in vivo to benzoylecgonine, ecgonine methyl esther, and norcocaine. The coingestion of cocaine and ethanol produces cocaethylene that can be detected in urine after 24 h. Because of the short half-life of cocaine, the analysis of metabolites in biological matrices is appropriate for monitoring cocaine abuse.
CE coupled to an ion-trap mass spectrometer has been evaluated for the analysis of cocaine and its main metabolites in human urine (benzoylecgonine, cocaethylene, anhydroecgonine, anhydroecgonine methyl ester, and ecgonine methyl ester) (36). The CE separation was performed in acidic conditions and obtained the same migration times for almost all compounds, which were identified with the MS detector. LODs in the range
100–250 ng/mL were found for all the analytes.
The limited sensitivity of the method was probably a result of the poor sample pretreatment. Urine samples were treated with acetonitrile and diluted with water before analysis, without any further clean-up or preconcentration step.
A sensitive method was proposed by Hezinova and colleagues (37) for the simultaneous determination of cocaine and its metabolites in spiked urine samples by CE coupled to electrospray ionization (ESI) and MS using a pressurized liquid junction nano-flow interface. The complete separation of cocaine, cocaethylene, benzoylecgonine, norcocaine, and ecgonine methyl ester was obtained in less than 15 min using an alkaline buffer (pH 9.5). For sensitivity enhancement, a field-amplified sample injection (FASI) on-column preconcentration was evaluated, achieving LOD values in the range 1.5–10 ng/mL. To further increase the method sensitivity, the spiked urine samples were subjected to an SPE procedure. With respect to the method described previously, the electrophoretic separation of norcocaine and benzoylecgonine, both characterized by the same molecular mass, a gain in method sensitivity of 25–50-fold was achieved.
The detection of cocaine in biological fluids using CE coupled to laser-induced fluorescence (LIF) and using a nucleic acid aptamer as affinity probe was presented (38). The presence of cocaine was observed by the variations in the fluorescence intensities of the free and complex forms of the aptamer. The developed method was applied to the analysis of cocaine in a bovine fetal serum, obtaining a LOD of 5 µM.
A fast and sensitive CE time-of-flight (TOF)-MS method for the screening of drugs of abuse (cocaine and methadone) in urine was developed by Kohler and colleagues (39). Analyses were performed using a simple urine dilution and on-line preconcentration approach with pH-mediated stacking, achieving LODs in the range 5–50 ng/mL for cocaine and its metabolites. The quantification of cocaine and methadone was performed in urine samples collected from drug consumers.
Analysis of Amphetamines
Amphetamines are strong stimulants of the central nervous system and are taken as recreational drugs. The abuse of these designer drugs can lead to hallucinations and psychosis, as well as dysphoria and depression. These synthetic compounds derive from the β-phenylethylamine core structure, chief among them are amphetamine, methamphetamine, and 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) (40). The quantification of these drugs and the determination of their metabolites in biological fluids obtained from suspected addicts is of great importance in clinical and forensic analysis.
A direct CE method with capacitively coupled contactless conductivity detection (C4D) for the simultaneous separation of seven amphetamine analogues as well as amphetamine, dextroamphetamine, methamphetamine, and MDMA was proposed (41). Electrophoretic parameters such as pH, concentration buffer, and cyclodextrin (CD) concentration were investigated to separate all the amphetamine-type substances. UV and C4D detectors were used simultaneously using the same electrophoretic conditions to obtain comparable sensitivities in the range of 1–4.4 µg/mL. The method was used for the analysis of street-grade ecstasy and dexamphetamine tablets.
Since amphetamine is a chiral drug, enantiomeric separation of amphetamine plays a fundamental role in the determination of synthetic pathway and for impurity profiling of the drug; dexamphetamine is used in the treatment of the attention deficit hyperactivity disorder (ADHD) of children and narcolepsy. Moreover, the (S)-enantiomer of dexamphetamine is a controlled substance in most European countries and the US. The enantiomeric purity of dexamphetamine and its potential impurities 1R,2S-(−)-norephedrine and 1S,2S-(+)-norpseudoephedrine was performed by using heptakis-(2,3-di-O-acetyl-6-O-sulfo)-β-CD (42). This CD resulted in the most suitable chiral selector among a series of charged and uncharged CDs.
In a further study, the simultaneous determination of the potential impurities, 1S,2S-(+)-norpseudoephedrine, 1R,2S-(-)-norephedrine, phenylacetone, phenylacetone oxime of dexamphetamine sulphate, and its stereoisomer levoamphetamine, was achieved by a CE method that used a dual CD system composed of sulfobutylether-β-CD and sulfated β-CD (43). The method allowed the quantification of impurities at the 0.05% level, in accordance with the previous study (42). Furthermore, this method allowed the CE separation of the E and Z stereoisomers of phenylacetone oxime, neutral impurities involved in the synthesis process of dexamphetamine.
As an alternative a CD-modified microemulsion electrokinetic method was proposed for the same analysis, where only sulfated β-CD was used as a chiral selector in combination with sodium dodecyl sulfate (SDS) (44). The validated method produced LOD values for all analytes between 0.05% and 0.2%, against a concentration of 3 mg/mL dexamphetamine sulfate.
Another dual CD system, including heptakis (2,6-di-O-methyl)-β-CD (DM-β-CD) and β-CD, was developed for the chiral separation of methamphetamine and its related compounds (45). To better control the electroosmotic flow and avoid the consequent analyte peaks shift, and improve the reproducibility of migration times for analytes by CE, the capillary wall was chemically modified with diol groups. Performing analyses in two different conditions, that is, both suppressing and promoting electroosmotic flow (EOF), the diol capillary gave better results in terms of reproducibility, compared with an untreated and a PVA capillary, even when used for the analysis of urine samples.
As mentioned previously, one advantage of reducing analytical techniques down to chip level is the portability of the instrument, directly into the place where a rapid screening is needed, such as clandestine laboratories. The suitability of this kind of lab-on-chip analyses was initially demonstrated for the analysis of biological molecules (46). In the optimized conditions, the separation of ephedrine, pseudoephedrine, and methamphetamine was obtained in less than 1 min and the analytical system was applied to different clandestine laboratory samples.
Analysis of New Psychoactive Substances and other Recreational Drugs
There has been a huge increase in the consumption of new psychoactive substances (NPSs) in recent years all over the world. Many new emerging drugs of abuse are marketed as legal highs, plant food, or bath salts, despite being labeled “not for human consumption” to circumvent drug abuse legislation (47–50).
The easy availability via the internet and at local “headshops” without any legal restriction, as well as the moderate costs, are the main reasons for this emerging phenomenon, with growing cases of acute intoxication and death.
The NPSs include synthetic cannabinoids, synthetic cathinones, phenetylamines, and piperazine derivatives. These designer drugs are synthetic compounds with a chemical structure slightly modified from the chemical structure of existing illicit drugs of abuse (for example, opioids, amphetamines, and marijuana) and provide hallucinogenic, psychotropic, and stimulating effects.
Despite recent work, the purity, pharmacokinetic, and toxicity profiles of these compounds are largely unknown. Conventional chromatographic approaches (HPLC, GC) hyphenated to MS have been evaluated to identify and quantify these substances and their metabolites in biological fluids (11,51). Up until now, few examples have been reported in the literature regarding the analysis of these compounds by miniaturized separation techniques. In the last decade, a new group of psychoactive substances, cathinone derivatives, have appeared on the market as recreational drugs. These synthetic compounds, also called beta-keto amphetamines, possess nearly the same stimulating effects as illicit amphetamines. In addition to the native cathinone, which is the constituent of the khat (Catha edulis) plant, used for centuries for its psychoactive effect, a wide range of derivatives have been synthesized (52).
Roda and colleagues (53) developed a simple and fast CE method for the determination of active principles of Catha edulis (cathinone, cathine, and phenylpropanolamine) and separated the three compounds and nicotinamide in 6 min. The electrophoretic method was validated following the guidance on validation published by the European Medicines Agency (53). LOD and LOQ values of 0.2 µg/mL and 0.4 µg/mL were achieved, respectively. Particular attention was paid to selecting the appropriate extraction solvent and the proper extraction method for the khat leaves. Maceration was considered the technique of choice for the extraction of khat alkaloids, using methanol as extraction solvent. The conservation of the vegetable material was evaluated, and confirmed that drying is the best way to preserve the active principles.
Similar to other drugs of abuse (amphetamines), the S (-) enantiomer of methcathinone is more potent than the R (+) antipode, therefore both the pharmacological and toxicological data of each stereoisomer are of importance both for legal and medical purposes. In addition, determination of the enantiomeric composition of those compounds yields information about the starting materials and laboratories of origin, enabling the tracking of the synthetic drugs.
A chiral CE method for the enantiomeric separation of a set of 19 cathinone derivatives was developed (54). Several neutral and charged CDs were investigated and negatively charged sulfated-β-CD was selected for the best resolution. In the optimized conditions, all the compounds, except methedrone, were resolved in less than 20 min.
Among the NPSs, synthetic cannabinoids have grown in popularity. The compounds are contained in herbal mixtures called “spices,” which are smoked as an apparently psychotropic legal alternative to cannabis. Most synthetic cannabinoids belong to the class of N-alkylindoles; some of these substances are 100 times more potent than cannabis (48).
The simultaneous separation of 12 synthetic cannabinoids and 9-Δtetrahydrocannabinol (Δ9-THC) in herbal blends by nano-LC was reported by Merola and colleagues (55). The method was developed through selecting the proper stationary phase and mobile phase composition. Under the optimized conditions, all of the compounds were resolved using an isocratic elution mode in less than 30 min, as illustrated in Figure 1. The nano-LC method was able to determine synthetic cannabinoids in herbal mixtures and had undergone a very simple sample pretreatment based on methanolic extraction. In addition, nano-LC–MS and nano-LC–MS2 experiments were performed for the characterization of the 12 synthetic cannabinoids, employing ion-trap MS by way of a nano-electrospray interface.

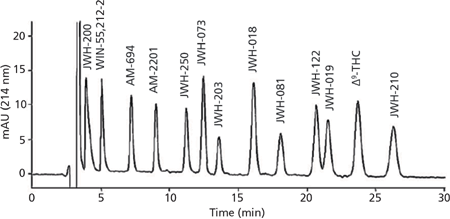
Figure 1: Nano-LC separation of synthetic cannabinoids and Δ9-THC using the optimum experimental conditions: mobile phase, acetonitrile/methanol/H2O/formic acid 69/5/25/1 (v/v/v/v); flow rate, 500 nL/min; concentration of all analytes 25 µg/mL and Δ9-THC 50 µg/mL. Adapted and reproduced with permission from Journal of Pharmaceutical and Biomedical Analysis 71, G. Merola, Z. Aturki, G. D'Orazio, R. Gottardo, T. Macchia, F. Tagliaro, and S. Fanali, Analysis of synthetic cannabinoids in herbal blends by means of nano-liquid chromatography, 45–53 (2012) © Elsevier.
The same class of psychoactive substances was analyzed in herbal blends by MEKC (56). The optimized MEKC method allowed the separation of 10 synthetic cannabinoids in less than 15 min. The method was validated and applied to the determination of synthetic cannabinoids in 15 different herbal blend samples that had been diluted before analysis. The value of the MEKC technique was also demonstrated by estimating the octanol-water partition coefficients (log P) of these compounds.
A rapid CE-UV method for the determination of three chlorophenylpiperazine isomers: 1-(2-chlorophenyl)piperazine (oCPP), 1-(3-chlorophenyl)piperazine (mCPP), and 1-(4-chlorophenyl)piperazine (pCPP) in illegal pills was proposed (57). To separate the isomers, several CD derivatives were investigated, and α-CD was selected. The presence of other candidate interferents (amphetamine, methamphetamine, 3,4-methylenedioxyamphetamine, 3,4-methylenedioxymethamphetamine, 3,4-methylenedioxy-N-ethylamphetamine, 1-[3-trifluoromethylphenyl]piperazine, and cocaine) as adulterants in pills was also examined. The proposed method was validated and provided LODs of 2.0 µg/mL (oCPP), 2.5 µg/mL (mCPP), and 3.5 µg/mL (pCPP). The analysis of 17 confiscated pills based mostly on mCPP was performed and identified other illicit drugs in the illegal samples, such as methamphetamine, MDMA, and cocaine.
In the case of new amphetamine-like designer drugs, the derivative compounds are characterized by the presence of one chiral centre, therefore the analysis of the enantiomeric composition could be very useful for investigative purposes, since the enantiomeric excess can provide important information about the synthesis pathway and about the laboratory of origin. In this respect, 13 synthesized amphetamine-derivates were enantiomerically separated and the influence of three different CD derivatives, namely, sulfated-β-CD, carboxymethyl-β-CD, and dimethyl-β-CD (58) were studied. Sulfated-β-CD gave the best results in terms of resolution and analysis time.
A well-known anesthetic drug used in veterinary medicine and in some surgical procedures for humans, ketamine is also abused as a recreational drug because it produces prolonged hallucination and delirium. As a result of its rapid metabolic degradation, a hair sample is preferred to investigate past drug consumption. An HPLC–chip-MS-MS method was developed for the simultaneous quantitative analysis of ketamine and its metabolite norketamine in human hair (59). It showed extremely high detection sensitivity at the femtogram level, and required only a few milligrams of hair sample.
Multidrug Analysis
In the competition to offer the most suitable analytical techniques, one of the main aims is to formulate a method that can provide rapid screening and quantification of as many compounds as possible, including those with completely different chemical and physical properties. This is particularly important in the forensic field because in many cases the type of drug present in seized materials or biological samples has to be assumed initially, before identification can be made. A metabolomic approach, based on the coupling of a separation technique with (MS-MS), is needed for the study of comprehensive drug metabolism and toxicity and complicated by the common use of drug cocktails (60).
The analysis of multiple drugs often involves mixtures of amphetamines, opiates, and hypnotic drugs, and their metabolites. Sweeping CE was used for the analysis of codeine, ketamine, methamphetamine, morphine, and benzodiazepines (BDZ)such as alprazolam and oxazepamand their metabolites in urine (61). A chemometric experimental design was used for the optimization of several parameters and to find the best compromise between an optimized separation of compounds and analysis time. A total of 14 compounds were separated in 26 min with detection limits in the 20–50 ng/mL range.
A similar approach, based on large volume sample stacking (LVSS)-sweeping CE, and developed with the help of interpretative strategies, was adopted for the same compounds, achieving LODs of 7.5–30 ng/mL and reducing analysis time to 10 min (62).
Conventional LLE was selected for the analysis of 12 drugs of abuse in vitreous humor samples, a less complex matrix (63). To increase sensitivity, a stacking strategy was performed by injecting a water plug prior to electrokinetic sample injection; LOD values of 1–5 ng/mL were achieved.
Sensitivity is the Achilles’ heel of miniaturized techniques, especially when UV detection is used, much research has been focused on the optimization of sample cleanup and preconcentration. Magnetic solid-phase extraction combined with FASS was proposed to enhance sensitivity in CE for the analysis of ketamine, amphetamines, and opiates, reaching limits of detection between 15 and 105 ng/mL (64).
Dispersive liquid–liquid microextraction (DLLME) was used for preconcentrating heroin, DL-methamphetamine, DL-3,4-methylenedioxymethamphetamine, and DL-ketamine. The technique, when combined with CE–UV, achieved LODs of 0.05-0.20 μg/L (65). The method was able to discriminate the enantiomers by the use of β-CD in the running buffer, and was applied to chiral separation and determination of studied illicit drugs on spiked samples, such as banknotes, kraft paper, plastic bags and silver paper, and produced good results.
DLLME combined with FASI was used to obtain low detection limits for a CE–UV method, which was developed for the analysis of MDMA, lysergic acid diethylamide (LSD), and phencyclidine (PCP) in human urine samples (66). Experimental design (central composite design) and the response surface methodology (RSM) were used for the optimization of the DLLME procedure. LODs of 1.00 ng/mL, 4.50 ng/mL, and 4.40 ng/mL were obtained for MDMA, PCP, and LSD, respectively.
A further way to increase sensitivity and specificity of CE lies in coupling it with MS. Electrospray ionization is preferred alongside this hyphenated method and several interfaces are available to allow the implementation of this dual system. However, some crucial parameters, such as the flow rate of the sheath liquid, the applied voltage, the composition, and the concentration of the buffer have to be carefully optimized. Even if the use of nonvolatile buffers is not recommended for MS coupling, they often offer higher efficiency and resolution compared with volatile buffers for CE separations. However, some papers have demonstrated the possibility of using nonvolatile buffers in CE–MS; a study was performed for the optimization of the CE–ESI–MS analysis of a mixture of forensic drugs (3,4-methylenedioxyamphetamine [MDA], MDMA, methadone, morphine, codeine, 6-monoacetylmorphine [6-MAM], and cocaine) and applied to the separation of drugs of abuse in real samples of hair, using a phosphate buffer (67). LOD values were as low as 0.01–0.008 ng/mg.
As well as increasing sensitivity, MS coupling is essential in metabolomic studies. CE coupled to TOF-MS was exploited for monitoring compliance to therapy during addiction treatments (68). Hair was used as the specimen because it allows an increased time-window for detection of drug metabolites, as opposed to blood or urine. In addition, collection is non-invasive and well accepted by patients.
DLLME combined with CE and TOF-MS was investigated for the detection of 30 toxicological compounds (amphetamines and their derivatives, opiates, cocaine and its metabolites, and pharmaceuticals) in urine samples; LODs down to sub-nanogram-per-milliter levels were achieved (69).
Multidrug analysis involves the use of other miniaturized techniques previously described in this review. A mixture of 10 illicit drugs was separated using CEC and a cyano stationary phase, which offered a better performance than the more commonly used C18 stationary phases for such basic compounds (70). On-line preconcentration by FASI was selected and achieved detection limits in the 5–12 ng/mL range. The developed method was applied to the analysis of urine samples, which had been subjected to a SPE procedure on strong cation-exchange cartridges.
Coupling with MS can also be extremely advantageous; however, it is even more complicated than CE and still requires improvement. An update on the work described above involved the hyphenation of a CEC system with an ion-trap MS system (71). The coupling with MS required the use of a volatile buffer and the adjustment of mobile phase composition. LOD values down to 0.78 ng/mL and 3.12 ng/mL were achieved, without affecting analysis time.
Another proposed method for the analysis of illicit drugs described the use of single drop microextraction (SDME) and open tubular capillary electrochromatography (OT-CEC), using multiwall carbon nanotubes (MWCTs) immobilized into a fused-silica capillary as the stationary phase (72). The use of SDME resulted in an enrichment factor of between 38- and 102-fold, depending on the drug, while the presence of MWCTs increased the selectivity and efficiency of the system.
Drugs of abuse and their metabolites were simultaneously measured by a microfluidic chip-based nano-HPLC coupled to MS-MS in the hair of drug abusers (73). The system integrated an enrichment column, an analytical column, and a nanospray tip and achieved chromatographic separation in 15 min, with LOD values from 0.1 pg/mg to 0.75 pg/mg.
Conclusions
Drug abuse is still a huge problem in society, and combines health and safety concerns with legal and forensic interests. Analytical chemistry plays a fundamental role in many of these areas because it allows the detection of drugs in seized materials, as well as in biological matrices, and consequently the study of pharmacokinetic and pharmacodynamic processes.
As well as established techniques, such as HPLC and GC, which are widely used in private and governmental laboratories, miniaturized analytical techniques are increasingly being used because of their reduced reagent consumption, lower waste disposal, fast analysis, and easy automation. CE is playing a prominent role, particularly in forensic analysis. However, miniaturized LC should be seen as a valid alternative to conventional chromatographic methods too. The works presented in this review support this statement.
References
(1) T. Ter Bogt, H. Schmid, S. Nic Gabhainn, A. Fotiou, and W. Vollebergh, Addiction101, 241–251 (2006).
(2) R.M. Eckersley, Drug Alcohol Rev.24, 157–163 (2005).
(3) L. Degenhardt and W. Hall, The Lancet379, 55–70 (2012).
(4) U.S. Department of Justice - National Drug Intelligence Center, Product No. 2011-Q0317-002.
(5) J.I. Khan, T.J. Kennedy, and D.R. Christian Jr., Basic Principles of Forensic Chemistry (Humana Press, Springer Science+Business Media, New York, USA, 2011).
(6) http://drugabuse.com/library/history-of-drug-abuse/
(7) K. Wada, Ann. N.Y. Acad. Sci.1216, 62–72 (2011).
(8) R. Room and P. Reuter, The Lancet379, 84–91 (2012).
(9) C. Carstairs, Drug Alcohol Rev.24, 57–65 (2005).
(10) C.S.J. Fazey, Int. J. Drug Policy14, 155–169 (2003).
(11) D. Favretto, J.P. Pascali, and F. Tagliaro, J. Chromatogr. A1287, 84–95 (2013).
(12) T.J. Raharjo and R. Verpoorte, Phytochem. Anal.15, 79–94 (2004).
(13) H.H. Maurer, Clin. Biochem.38, 310–318 (2005).
(14) C. Cruces-Blanco, L. Gámiz–Gracia, and A.M. García-Campaña, Trends Anal. Chem.26, 215–226 (2007).
(15) C. Cruces-Blanco and A.M. García-Campaña, Trends Anal. Chem.31, 85–95 (2012).
(16) E. Al-Hetlani, Electrophoresis34, 1262–1272 (2013).
(17) Z. Aturki, A. Rocco, S. Rocchi, and S. Fanali, J. Pharm. Biomed. Anal.http://dx.doi.org/10.1016/j.jpba.2014.03.041, in press (2014).
(18) A.E. Schwaninger, M.R. Meyer, and H.H. Maurer, J. Chromatogr. A1269, 122–135 (2012).
(19) I.S. Lurie, R.F.X. Klein, T.A. Dal Cason, M.J. LeBelle, R. Brenneisen, and R.E. Weinberger, Anal. Chem.66, 4019–4026 (1994).
(20) I.S. Lurie, P.A. Hays, A.E Garcia, and S. Panicker, J. Chromatogr. A1034, 227–235 (2004).
(21) Z. Zhang, B. Yan, K. Liu, Y. Liao, and H. Liu, Electrophoresis30, 379–387 (2009).
(22) J. Rittgen, M. Pütz, and R. Zimmermann, Electrophoresis33, 1595–1605 (2012).
(23) J. Sheng, Q. Ping, J. Lei, H. Ju, C. Song, and D. Zhang, Anal. Lett.45, 652–664 (2012).
(24) I. Botello, F. Borrull, C. Aguilar, and M. Calull, Electrophoresis33, 528–535 (2012).
(25) I. Botello, F. Borrull, M. Calull, C. Aguilar, G.W. Somsen, and G.J. de Jong, Anal. Bioanal. Chem.403, 777–784 (2012) .
(26) Y.H. Tak, J. Sastre Toraño, G.W. Somsen, and G.J. de Jong, J. Chromatogr. A1267, 138–143 (2012).
(27) P. Meng, Y. Wang, and L. Meng, Anal. Methods4, 3695–3700 (2012).
(28) Y. Iwamuro, R. Iio-Ishimaru, S. Chinaka, N. Takayama, and K. Hayakawa, Biomed. Chromatogr.26, 1452–1456 (2012).
(29) J. Li and Y. Jiang, Biomed. Chromatogr.24, 186–194 (2010).
(30) J. Roscher, T. Nils Posch, M. Pütz, and C. Huhn, Electrophoresis33, 1567–1570 (2012).
(31) J. Sáiz, T. Duc Mai, M. López López, C. Bartolomé, P.C. Hauser, and C. García-Ruiz, Sci. Justice53, 409–414 (2013).
(32) J.J. Platt, Cocaine Addiction, Theory, Research and Treatment (Harvard University Press, Cambridge, MA, USA, 1997).
(33) R.J. Devlin and J.A. Henry, Crit. Care12, 202–209 (2008).
(34) J. Glauser and J.R. Qeen, J. Emerg. Med.32, 181–186 (2007).
(35) C.M. Nzerue, K. Hewan-Lowe, and L.J. Riley Jr., Am. J. Kidney Dis.35, 783–795 (2000).
(36) J.L. da Costa, F.G. Tonin, L.A. Zanolli, A.A.D. Chasin, and M.F.M. Tavares, Electrophoresis30, 2238–2244 (2009).
(37) V. Hezinová, Z. Aturki, K. Klepárník, G. D'Orazio, F. Foret, and S. Fanali, Electrophoresis33, 653–660 (2012).
(38) Q.P. Deng, C. Tie, Y.L. Zhou, and X.X. Zhang, Electrophoresis33, 1465–1470 (2012).
(39) I. Kohler, J. Schappler, and S. Rudaz, Anal. Chim. Acta780, 101–109 (2013).
(40) M. Carvalho, H. Carmo, V. M. Costa, J P. Capela, H. Pontes, F. Remião, F. Carvalho, and M. de Lourdes Bastos, Arch Toxicol.86, 1167–1231 (2012).
(41) R. Epple, L. Blanes, A. Beavis, C. Roux, and P. Doble, Electrophoresis31, 2608–2613 (2010).
(42) N.G. Kokiashvili, S.Wongwan, C. Landgraf, K. Michaelis, M. Hammitzsch-Wiedemann, and G.K.E. Scriba, J. Pharm. Biomed. Anal.50, 1050–1053 (2009).
(43) S. Wongwan, B. Sungthong, and G.K.E. Scriba, Electrophoresis31, 1475–1481 (2010).
(44) S. Wongwan and G.K.E. Scriba, Electrophoresis31, 3006–3011 (2010).
(45) Y. Iwamuro, R. Iio-Ishimaru, S. Chinaka, N. Takayama, S. Kodama, and K. Hayakawa, Forensic Toxicol.28, 19–24 (2010).
(46) A. Lloyd, M. Russell, L. Blanes, P. Doble, and C. Roux, Forensic Sci. Int.228, 8–14 (2013).
(47) M.E. Musselman and J.P. Hampton, Pharmacotherapy doi:10.1002/phar.1424, in press (2014).
(48) I. Vardakou, C. Pistos, and C. Spiliopoulou, Toxicol. Lett.197, 157–162 (2010).
(49) J.B. Zawilska and J. Wojcieszak, Forensic Sci. Int.231, 42–53 (2013).
(50) C.L. German, A. E. Fleckenstein, and G.R. Hanson, Life Sciences97, 2–8 (2014).
(51) M.A. Elsohly, W. Gul, A.S. Wanas, and M.M. Radwan, Life Sciences97, 78–90 (2014).
(52) J.P. Kelly, Drug Test Anal.3, 439–453 (2011).
(53) G. Roda, V. Liberti, S. Arnoldi, A. Argo, C. Rusconi, S. Suardi, and V. Gambaro, Forensic Sci. Int.228, 154–159 (2013).
(54) S. Mohr, S. Pilaj, and M.G. Schmid, Electrophoresis33, 1624–1630 (2012).
(55) G. Merola, Z. Aturki, G. D'Orazio, R. Gottardo, T. Macchia, F. Tagliaro, and S. Fanali, J. Pharm. Biomed. Anal.71, 45–53 (2012).
(56) R. Gottardo, A. Bertaso, J. Pascali, D. Sorio, G. Musile, E. Trapani, C. Seri, G. Serpelloni, and F. Tagliaro, J. Chromatogr. A1267, 198–205 (2012).
(57) J. Siroká, D.N. Polesel, J.L. Costa, R. Lanaro, M.F.M. Tavares, and M. Polášek, J. Pharm. Biomed. Anal.84, 140–147 (2013).
(58) L. Burrai, M. Nieddu, M.A. Pirisi, A. Carta, I. Briguglio, and G. Boatto, Chirality25, 617–621 (2013).
(59) K.Y. Zhu, K. Wing Leung, A.K.L. Ting, Z.C.F. Wong, Q. Fu, W.Y.Y. Ng, R.C.Y. Choi, T.T.X. Dong, T. Wang, D.T.W. Lau, and K.W.K. Tsim, Forensic Sci. Int.208, 53–58 (2011).
(60) A.L. Patton, K.A. Seely, K.C. Chimalakonda, J.P. Tran, M. Trass, A. Miranda, W.E. Fantegrossi, P.D. Kennedy, P. Dobrowolski, A. Radominska-Pandya , K.R. McCain, L.P. James, G.W. Endres, and J.H. Moran, Anal. Chem.85, 9390–9399 (2013).
(61) J.-Feng Chiang, Y.-T. Hsiao, W.-K. Ko, and S.-M. Wu, Electrophoresis30, 2583–2589 (2009).
(62) Y.-H. Ho, C.-C. Wang, Y.-T. Hsiao, W.-K. Ko, and S.-M. Wu, J. Chromatogr. A1295, 136–141 (2013).
(63) J.L. Costa, A. Ribeiro Morrone, R. Ribeiro Resende, A. Aparecida da Matta Chasin, and M.F.M. Tavares, J. Chromatogr. B945–946, 84–91 (2014).
(64) M.-L. Chen, L.-L. Suo, Q. Gao, and Y.-Q. Feng, Electrophoresis32, 2099–2106 (2011).
(65) L. Meng, B. Wang, F. Luo, G. Shen, Z. Wang, and M. Guo, Forensic Sci. Int.209, 42–47 (2011).
(66) D. Airado-Rodríguez, C. Cruces-Blanco, and A.M. García-Campaña, J. Chromatogr. A1267, 189–197 (2012).
(67) R. Gottardo, I. Mikšík, Z. Aturki, D. Sorio , C. Seri, S. Fanali, and F. Tagliaro, Electrophoresis33, 599–606 (2012).
(68) R. Gottardo, A. Fanigliulo, D. Sorio, E. Liotta, F. Bortolotti, and F. Tagliaro, Forensic Sci. Int.216, 101–107 (2012).
(69) I. Kohler, J. Schappler, T. Sierro, and S. Rudaz, J. Pharm. Biomed. Anal.73, 82–89 (2013).
(70) Z. Aturki, G. D'Orazio, S. Fanali, A. Rocco, F. Bortolotti, R. Gottardo, and F. Tagliaro, J. Chromatogr. A1216, 3652–3659 (2009).
(71) Z. Aturki, G. D'Orazio, A. Rocco, F. Bortolotti, R. Gottardo, F. Tagliaro, and S. Fanali, Electrophoresis31, 1256–1263 (2010).
(72) P.W. Stege, A.V. Lapierre, L.D. Martinez, G.A. Messina, and L.L. Sombra, Talanta86, 278–283 (2011).
(73) K.Y. Zhu, K. Wing Leung, A.K.L. Ting, Z.C.F. Wong, W.Y.Y. Ng, R.C.Y. Choi, T.T.X. Dong, T. Wang, D.T.W. Lau, and K.W.K. Tsim, Anal. Bioanal. Chem.402, 2805–2815 (2012).
Zeineb Aturki received her degree in industrial chemistry in 1992 at the University “La Sapienza” in Rome. In 1994, she won a grant and began her research experience at the Istituto di Cromatografia, Consiglio Nazionale delle Ricerche (CNR). Since 2001, Zeineb has worked as an investigator in a permanent position at the Istituto di Metodologie Chimiche, Consiglio Nazionale delle Ricerche (CNR). Her research fields include: electromigration and chromatographic techniques including micro and nano-liquid chromatography, capillary electrophoresis, and capillary electrochromatography; hyphenation of the miniaturized separation techniques with mass spectrometry; analysis of compounds of food, pharmaceutical, and forensic interest; and analysis of chiral compounds, with the investigation of new chiral selectors and chiral stationary phases for the separation of optical isomers.
Anna Rocco graduated in pharmaceutical chemistry and technologies from the University of Rome “La Sapienza,” Faculty of Pharmacy and Medicine, in 2003. She obtained her PhD degree in chemistry, physical science area, at Vytautas Magnus University (Kaunas, Lithuania), in 2013. Since 2004, she has been involved in the research of separation science at the Istituto di Metodologie Chimiche, Consiglio Nazionale delle Ricerche (CNR), in Rome, under the supervision of Dr. S. Fanali. Her main interests are related to the development of analytical methods for the determination of compounds of pharmaceutical, food, and environmental interest (including chiral separations) by means of capillary electrophoresis, capillary electrochromatography, and nano-liquid chromatography coupled with UV or MS detection.
Salvatore Fanali is a senior researcher (Research Director) at the Institute of Chemical Methodologies, Italian National Research Council in Monterotondo (Rome, Italy). He received the title of Dr. in chemistry at Rome University “La Sapienza” and a PhD in analytical chemistry at Comenius University Bratislava (Slovak Republic). His research activity is focused mainly on the development of new miniaturized instrumentation and methods including CE, CEC, capillary, and nano-LC-all hyphenated with MS.




















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

