How Raw Are Your Data — 2012?


The results of a recent survery on stationary phases used in high performance liquid chromatography (HPLC) are revealed.
This column revisits the topic of raw data and electronic records for a chromatography data system (CDS) in light of recent updates in regulations and guidance issued by regulatory agencies and industry bodies. We look at the ways to define what raw data are for a CDS and pose the question: Is it still possible to define paper records as chromatographic raw data?
In this Questions of Quality column I want to return to a topic that I have discussed twice before in the past few years: What are the raw data and/or electronic records for a chromatography data system (CDS)? In December 1996 I published the first Questions of Quality on raw data (1) and, with the timing that snatched defeat out of the jaws of victory, I was in print before the final rule on electronic records and electronic signatures was issued by the FDA in March 1997 (2). Therefore in 2000, I returned to the subject of raw data in light of industry and my experience with Part 11 to discuss and define the scope of electronic records that you could have with a CDS (3). More recently, in 2011, there was the revision of EU GMP Annex 11 on computerized systems (4) and Chapter 4 on documentation (5) that I reviewed in this column last year (6). Although the Questions of Quality column focused on Annex 11, there were significant changes to Chapter 4 on documentation that I want to focus on in this column. This is the new requirement to define raw data for systems involved in making quality decisions (5). As a CDS will be used to release raw materials, finished products and stability testing, it will be involved in making quality decisions and the raw data needs to be defined. Therefore what should we do for a CDS? Has anything changed since I wrote the first article on this topic over fifteen years ago?
To give you an idea of what we will be discussing I have presented the regulations, regulatory guidance and industry guidance documents to be discussed in Figure 1. First we'll look back and I'll give you a summary of the points from the Questions of Quality columns from 1996 and 2000 (1, 3), the latter article being greatly influenced by the publication of 21 CFR 11 regulations (2). Then we'll begin by looking at the FDA Guidance on Part 11 Scope and Application published in 2003 (7), which was expanded with the FDA interpretation published in 2004 (8) for electronic versus paper records for a CDS.
Next, we will look in more detail at the specific EU GMP requirements for raw data from the new version of Chapter 4 on documentation (5). However, as Chapter 4 does not give a definition of raw data, we will look at the definitions in the US GLP regulations (9) and a Swiss GLP Guidance (10). Finally, we'll end with a look at what is said in the FDA's Compliance Programme Guide 7346.832 on Pre-Approval Inspections (11) and the second edition of the GAMPGood Practice Guide: RiskBased Approach to GxP Compliant Laboratory Computerized Systems (12).


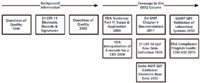
Figure 1: Outline of this month’s column on raw data.
QOQ 1996: In The Beginning
In the pre-Part 11 world, of my 1996 Questions of Quality article on raw data (1) I drew two main conclusions (1):
1. Electronic data files were preferable to paper printouts from a CDS.
2. Many laboratory practices involving electronic raw data were unacceptable with informally documented procedures for the use and maintenance of a CDS.
The problem in 1996 was that there were no regulations that stated what needed to be done; interpretations of the recommendations were based on old ways of working with paper that were extrapolated to the computerized systems of the time and in essence defined raw data as paper records.
To come to these conclusions I drew on the definition by Furman et al. (13) that "raw data were all those (e.g. records or files, my addition) that could be saved and accessed later." The authors, who were members of the FDA at the time their paper was published, pointed out that chromatographers should consider the following scenario: two small peaks are detected eluting ahead of a peak of interest which are ignored in the current method. Later evidence finds that these are minor compounds which are toxic and the organization needs to know how much of these compounds was present in all batches of material analysed in the past year. The authors then posed the question which of the following is preferable: reanalysing all batches of material or retrieving the old files of electronic raw data and re-integrate them? To Furman et al., their bottom line was very conservative: you should save all raw data, including the data files slices and the methods used to acquire the data, fit baselines and calculate results (13).
The second main reference was the paper by BARQA (British Association of Research Quality Assurance) (14) that outlined that raw data had four key factors:
- Original records or copies of original observations are required.
- Recorded directly, promptly, accurately, legibly and indelibly with observer identified. Raw data generated by direct input should be identified by the individual responsible for data entry.
- Changes to raw data should not obscure the original entries.
- There was a wide range of media that could be defined as raw data.
The advantages of defining electronic raw data are that storage is compact and efficient, and, like the argument from Furman, the data are available for further processing and analysis. Data can be copied or backed up with ease to provide duplicates that give increased security against loss or damage, providing it is documented and the copying is authenticated. That's where I left the raw data debate at the end of 1996.
QOQ 2000: The Bottom Line: Use Electronic Records?
When I revisited the raw data debate in 2000, what had changed? The publication of 21 CFR 11 that defined an electronic record as:
Any combination of text, graphics, data, audio, pictorial or other information representation in digital form that is created, modified, maintained, archived, retrieved or distributed by a computer system (2).
Furthermore, there was the need for efficient and effective archive and restore procedures for data systems in the chromatography laboratory as the regulations calls for:
Protection of records to enable their accurate and ready retrieval throughout the records retention period (§11.10c) (2).
So, after a discussion about the benefits of electronic records, I came to the following series of questions to help define what the electronic records were for any chromatographic data system. I stated that the key to success was to look carefully at your CDS application, know how it was used and the chromatographic instruments controlled by the system and then to consider the following questions:
1. Which files in your data system are needed to set up the system to acquire data and control the equipment? These will include any methods and modifications specifically for that run and the sequence of samples that include the sample identities, volumes, dilution factors and any post run calculations.
2. Are all the required files on the same or separate computers? If the latter, how will you be able to archive them efficiently?
3. Are any system suitability test and associated chromatographic data files available to demonstrate that the chromatograph and method was within specification at the time of the analysis?
4. Which data files were produced from each run? Are they correctly labelled and cross-referenced to the specific version of the other files used to interpret them?
5. What happens with re-injected samples: are the original files overwritten or do you have a second version of the file?
6. Which files are needed to produce results from the run? Here you'll need to think in more detail about how you interpret the data files, interpret the peaks (including any manual override from the automatic operation of the system), check the system suitability results, calibrate the run with the method, calculate the results in the unknowns, apply any correction factors or post-run calculations, approve the results and report them. Don't forget the files from the repeat samples or any dilutions.
7. Have you considered all eventualities, such as more esoteric detectors, e.g.. diode array detectors (DAD) or mass spectrometers with or without spectral libraries.
8. Have you included the audit trail entries for the run as electronic records?
9. Check that if you can archive all these files you can restore them as well.This is a two-way process. Remember that the archive medium you are starting with now may not be the one at the end of your record retention period.
So, by a series of simple questions, you can begin to define the electronic process that is used by the CDS to identify the electronic records used or created during an analysis – then all you need to do is document it. The column also gave a schematic of the electronic records for most CDS systems used in regulated environments.
Updating and Extending the Raw Data Debate
The introductory sections have summarized the debate I had in the first two Questions of Quality columns discussing what constitutes raw data. Now I want to bring the discussion up to date and widen the debate. To do this I will start with the FDA Guidance for Industry on Part 11 Scope and Application (7) that was published in 2003 at the start of the FDA's initiative on GMP's for the 21st century and take us through to the current situation as shown in Figure 1.
Exhibit 1: FDA Part 11 Guidance
In 2003, the FDA issued first a draft and then a final version of the Part 11 Scope and Application guidance for industry (7). For an FDA guidance to go from draft to final version in seven months is the regulatory equivalent of exceeding the speed of light. In this guidance document the FDA went back to basics for the interpretation of Part 11. Firms must understand the existing GLP or GMP predicate rule and interpret accordingly to see if Part 11 applies. To help the debate, under the section discussing Definition of Part 11 Records, the following scenarios were identified that could be applicable to laboratory records (7):
- Part 11 applies to records that are required to be maintained under predicate rule requirements and that are maintained in electronic format in place of paper format. Put simply, if you are working electronically in a regulated laboratory Part 11 applies to your activities.
- Part 11 also applies to records that are required to be maintained under predicate rules, that are maintained in electronic format in addition to paper format, and that are relied on to perform regulated activities. Even though you print out and sign the paper, if you use the electronic records to make decisions then Part 11 applies to the system.
- Part 11 also applies to electronic signatures that are intended to be the equivalent of handwritten signatures, initials and other general signings required by predicate rules. If any records are signed electronically then Part 11 applies (well it would, wouldn't it?).
However, the guidance also stated:
On the other hand, when persons use computers to generate paper printouts of electronic records, and those paper records meet all the requirements of the applicable predicate rules and persons rely on the paper records to perform their regulated activities, FDA would generally not consider persons to be "using electronic records in lieu of paper records" under §§ 11.2(a) and 11.2(b). In these instances, the use of computer systems in the generation of paper records would not trigger Part 11 (7).
This is the typewriter interpretation or excuse, whereby a computer is just a means of printing out data. However, this argument fails to consider that virtually any computerized system in the laboratory, including an analytical balance, can actually perform calculations, be used to interpret the original data file and/or store electronic records. Most people reading the Part 11 guidance section tended to think that as they had used hand-signed paper printouts to comply with the regulations in the past that it would continue to be accepted to state that Part 11 would not apply to their system. Therefore, our raw data are the signed paper printouts and not the electronic records that generated them. This is the regulatory equivalent to limbo dancing under toilet doors.
What has not been considered in this approach is that the Part 11 guidance required a fundamental reinterpretation of the predicate rule requirements from the electronic record perspective and not that of paper. Even today, I meet chromatographers working in regulated laboratories who still insist, or their Quality Assurance Department does, on defining the raw data from chromatographic data systems as paper. Polite words fail me at this point.
The final section on the narrow interpretation of Part 11 finished with a statement that for each record required by the predicate rule the FDA recommended the regulated organisation document if paper or electronic records were used for regulated activities (7). This is important because it leads directly to the new requirement under the new version of EU GMP Chapter 4 to define records upon which quality decisions are made as raw data (5) that we shall encounter later in this column.
Exhibit 2: The Empire Strikes Back
In 2004, the FDA issued more detailed guidance aimed at CDSs used in Quality Control laboratories working to GMP. Although the principles that were outlined can be applied to virtually any laboratory computerized system used today. This was published on the FDA web site under the concise title: Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance — Records and Reports (8). The FDA web page states that this was issued in 2004, this did not come to prominence until last year and I was not aware of this gem until there was a rearrangement of the FDA web site. Item 3 on the web page is crucial to our discussion of how raw are your data (8), as it is a question concerning the interpretation of the GMP predicate rule (15) and the applicability of Part 11 (2) to chromatographic data systems.
The web page posed the question "How do the Part 11 regulations and predicate rule requirements for GMP (15) apply to the electronic records created by computerized laboratory systems and the associated printed chromatograms that are used in drug manufacturing and testing?" For computerized laboratory system read chromatographic data system. The FDA stated that "Some in industry misinterpret lines 164 to 171 (I have quoted them under Exhibit 1 above) from the Part 11 Guidance (7) to mean that in all cases paper printouts of electronic records satisfy predicate rule requirements in 21 CFR Part 211." This reiterates my comment above that this is an abuse of the typewriter interpretation as the guidance states that paper printouts are ONLY acceptable if they comply with the applicable predicate rules. Therefore, the key to the debate is what do the predicate rules state and how should they be interpreted for a CDS?
The FDA then comment that for a CDS and other computerized systems used in a Quality Control laboratory involving user inputs, outputs, audit trials, etc., there are two clauses from the GMP regulations applicable for the interpretation of paper versus electronic raw data debate. These are §211.68 and §211.180(d) (15).
- 21 CFR 211.180(d) states that manufacturing records must be retained "either as original records or true copies such as photocopies, microfilm, microfiche or other accurate reproductions of the original records." This clause shows how old the US GMP is because it mentions microfilm and microfiche. The regulation has not been updated since it was issued 1978 and is firmly grounded in a cellulose world.
- 21 CFR 211.68 further states that: "Hard copy or alternative systems, such as duplicates, tapes or microfilm, designed to assure that backup data are exact and complete and that it is secure from alteration, inadvertent erasures, or loss shall be maintained".
The FDA then makes the following statement which is reproduced below in its entirety; I have just added the bullet points to aid readability and understanding:
- The printed paper copy of the chromatogram would not be considered a "true copy" of the entire electronic raw data used to create that chromatogram, as required by 21 CFR 211.180(d).
- The printed chromatogram would also not be considered an "exact and complete" copy of the electronic raw data used to create the chromatogram, as required by 21 CFR 211.68.
- The chromatogram does not generally include, for example, the injection sequence, instrument method, integration method or the audit trail, of which all were used to create the chromatogram or are associated with its validity.
- Therefore, the printed chromatograms used in drug manufacturing and testing do not satisfy the predicate rule requirements in 21 CFR Part 211. The electronic records created by the computerized laboratory systems must be maintained under these requirements.
Why this lengthy debate on this point? The reason is that electronic records contain much more information than the corresponding paper printouts of the same chromatographic run. Consider an Excel file as a simple example; under the properties tab there is information about the file creation and printing dates which a typical printout does not contain unless specifically configured to do so. In this simple case the electronic record contains more information than the paper one. Go further to a CDS analytical run, who prints out the audit trail? However, the audit trail contains critical information to demonstrate the integrity of the analytical run and the conversion of the raw data files into the final reportable value and also identifies critical operator interactions that are have interpreted and modified the data and carried out calculations. Therefore, the electronic records from a CDS contain much more that just the paper printout and this is why the FDA want electronic records as noted in the last bullet point above: printed chromatograms do not satisfy the predicate rule requirements in 21 CFR 211.
In summary, a paper printout does not meet the requirements of the predicate rule and the CDS must therefore be considered either a hybrid system (electronic records with signed paper printouts) or a fully electronic system. Regardless of the way the CDS is used (hybrid or homogeneous system), the electronic records must be maintained and are key to meeting the predicate rule requirements.
However, I think that the FDA have missed a trick as a key regulatory requirement for the Quality Control laboratory is §211.194(a) (15) that states that "Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards". If you use the argument that your laboratory records are paper then you do not have a leg to stand on as paper records can never be complete. Note, although the FDA discusses mainly CDS systems, it does say at the start of the answer section that this discussion also includes other laboratory computerized systems (8).
Exhibit 3: New Requirement to Define GMP Raw Data
The new version of EU GMP Chapter 4 on documentation (5) became effective on 30 June 2011 and I reviewed this document together with the new version of Annex 11 in this column last year (6). The Principle of Chapter 4 lists the different types of documents expected in a GMP environment and makes two basic statements that are important to our debate:
1. Documentation may exist in a variety of forms, including paper-based, electronic or photographic media.
2. The term 'written' means recorded, or documented on media from which data may be rendered in a human readable form (5).
It is important to realize that in Chapter 4 the term "documentation" has a very wide scope, including electronic records. Also the definition of written is very wide and is independent of the medium that is used to record or document data and information. In fact this definition is so well written that even if a currently unknown medium is invented, developed and you use it to record regulated data, it automatically comes under the scope of Chapter 4.
The documents that are of most interest in our debate are records that are the result of following an instruction, for example, an analytical method. A CDS will generate electronic files during an analysis that fall under the Chapter 4 definition of records and there is a specific requirement that is crucial to the laboratory:
Records include the raw data which is used to generate other records. For electronic records regulated users should define which data are to be used as raw data. At least, all data on which quality decisions are based should be defined as raw data (5).
This is one of the major impacts of Chapter 4, the requirement to define the raw data in GMP regulated activities used to make quality decisions including paper, hybrid and electronic records. Therefore, electronic records which are used to make quality decisions should be defined as raw data. Moreover, if you convert the raw data to generate other records such as a dissolution profile using, say, a spreadsheet programme, these additional records and the printout are raw data and should also be defined. You will, of course, realize that when a regulation says "should" it really means "must".
Also according to Chapter 4, raw data is used to generate other records but does not go into any further details. Please put this topic on the back burner as we will return to this when we discuss the Swiss AGIT guidance document (10) later in this column.
In Chapter 4 clause 4.1 it states that all types of documents should be defined and adhered to and this applies to all media types. This clause then discusses hybrid and homogeneous documents as follows (5):
Many documents (instructions and/or records) may exist in hybrid forms, i.e. some elements as electronic and others as paper based. Relationships and control measures for master documents, official copies, data handling and records need to be stated for both hybrid and homogenous systems. Appropriate controls for electronic documents such as templates, forms and master documents should be implemented. Appropriate controls should be in place to ensure the integrity of the record throughout the retention period.
So, let us dissect this section in a little more detail. Regardless of the fact that a record from a CDS is homogeneous (fully electronic) or hybrid (electronic with a signed paper printout) the control mechanisms for these records need to be defined, documented and implemented under Chapter 4 regulations (5). One key requirement that both the FDA and Europeans agree on is record or data integrity: what controls are needed to ensure that the record is true, complete and accurate? A typical response from the regulator is "appropriate" – therefore more critical records need more stringent controls than non-critical ones. This has been discussed in some detail in the GAMP Good Practice Guide on Part 11 Electronic Records and Signatures compliance (16) and therefore I will not discuss this topic further here.
Documentation in the form of records is close to the definition of electronic record in 21 CFR 11 (2, 7) so care needs to be taken when discussing the term "documentation" in a European context as this can refer to electronic records. However, the big problem is that the term raw data is not defined in EU GMP Chapter 4. How can we get a regulatory definition of this term? Here's where we cross disciplines and look back in time so that we can go forward with this debate.
Exhibit 4: Back to the Future
If we look in Good Laboratory Practice regulations we can find the definition of the term raw data and we can start to interpret it for any regulated laboratory. The US GLP regulation, which has been effective since 1978, defines the term raw data in section §58.3(k) (9) and is reproduced below:
Raw data means any laboratory worksheets, records, memoranda, notes, or exact copies thereof, that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study. In the event that exact transcripts of raw data have been prepared (e.g., tapes which have been transcribed verbatim, dated and verified accurate by signature), the exact copy or exact transcript may be substituted for the original source as raw data. Raw data may include photographs, microfilm or microfiche copies, computer printouts, magnetic media, including dictated observations, and recorded data from automated instruments.
Some of the conclusions we can draw from this definition for a chromatography data system are that raw data are:
- Original observations. Raw data are the electronic files produced by the CDS and not the paper printout. The reason is similar to the FDA argument earlier in this column (8) that you need the sequence file, weights, dilution factors, purities, raw data, instrument control parameters, processing method and audit trail files before a paper or electronic report can be produced.
- Necessary for the reconstruction of the study report. A study report cannot be reconstructed from a paper printout as there is far more information contained in the electronic raw data than will ever be contained in a paper printout as we have discussed earlier under Exhibit 2. Moreover, everybody forgets about the audit trail. This is a critical component of data integrity of both hybrid and homogeneous computerized systems and is necessary for the reconstruction of any study report to identify who did what and when. If you are working in a GMP laboratory then the equivalent for a study report is a certificate of analysis, method development report, validation report or stability study report.
- Exact copies of original observations. These are the backup tapes or disk-to-disk copies of the electronic records created by the CDS and are essential if there is a problem with the computer hardware. In the laboratory, paper copies are verified by the person making the copies. The electronic equivalent of this will be validation that backup works and records that the backup procedure has been carried out daily and that there have been no problems with the process when the backup logs have been reviewed.
- Can be in any format or any media. This is similar to the definitions of record in Chapter 4 (5) and electronic record in Part 11 (2)
- Can be produced by automated instruments. Of course, our CDS is automated and produces raw data and all files must be retained and maintained as part of the overall records. The scope of this includes the chromatographic files, plus the associated metadata used to acquire the individual runs, interpret the data files and generate the report used to make decisions about the study or analytical samples, plus the audit trail. All of these files are raw data and are necessary for reconstruction of the study.
Therefore from a 30-year-old definition of raw data we can derive requirements applicable to electronic records generated by a CDS today. There is a similar definition of raw data in OECD GLP (17) which is used within Europe and several other countries that can be interpreted in the same way as US GLP that we have just completed above. However, we also need to consider the detail of the interpretation of the chromatographic files and now we move back into this century to another GLP regulatory guidance document, this time from Switzerland.
Exhibit 5: Swiss Electronic Raw Data Guidance
The Swiss AGIT (AGIT is the German abbreviation of the Working Group for Information Technology) group has produced an interesting guidance document that contributes to our debate. The AGIT are a group of Swiss GLP inspectors and industry experts working together to provide guidance for GLP regulated laboratories. They have produced a series of publications over the past 10 years and one that is the most relevant is entitled Guidelines for the Acquisition and Processing of Electronic Raw Data in a GLP Environment (10).
We will discuss the main points in this document from the perspective of the raw data debate. To begin, the guidance defines electronic raw data as:
Original test facility records generated by means of computerized systems and stored on digital media. In a broader sense this may include data processed subsequently, and stored on digital media, which are necessary for reconstruction and evaluation of the final results (10).
So we have two concepts in this definition:
- The initial capture of the raw data by a computerized system and this will be the initial acquisition of the data
- The subsequent interpretation of the electronic data, which in the case of a CDS will be the integration of the chromatograms of standards, blanks, quality controls and samples, to produce the reportable values or final results from an analytical run.
Second, the document then breaks down electronic raw data into various components as follows:
Electronic raw data are considered as the data themselves and their related metadata.
The data represent the core data elements (measured values), whereas metadata are considered as the attributes of the measured values (e.g. study number, time, sample identification) and technical properties (e.g. field properties, table relationships, keys etc.).
Additionally, all changes to electronic raw data have to be recorded in an audit trail specifying the original and modified data, the reason for the change, the date and time and the identity of the person changing the data. The processing of electronic raw data such as integration, calibration and calculation should be described by the process itself including processing parameters, equations and statistical methods. Intermediate results obtained during the data evaluation are not subject to audit trail and they do not necessarily have to be stored and maintained. However, the process finally applied and the corresponding results should be preserved (10).
Did you understand this section? No, I thought not, as this is written from an information technology perspective that confuses the message that it is trying to send. Perhaps a better approach to explain what this section means for a CDS is the following because it is crucial to the raw data debate:
- The chromatographic raw data file is the core data element or measured values described above, that is, the detector time slices. On its own a CDS data file (i.e. the chromatogram) is useless as there is no contextual data to say what it is or how it can be interpreted. For example, what method was used to control the instrument, how were the data acquired and what is the identity of each injection in the analytical run?
- To interpret each raw data file correctly requires the file to be put in context: therefore the instrument control file, an appropriate data acquisition method and sequence file define the instrument conditions, chromatographic method (e.g. data acquisition as well as the peak identification windows) and sample type to allow the correct interpretation of the data file. These are the metadata; the problem is that there is not a good definition of this word (e.g. data about data). However, without the metadata the data file is useless as it cannot be interpreted and is just the confidential questionnaire. This is shown in Figure 2 on the left hand side of the figure as the acquisition phase of an analysis.
- After the analysis the raw data files are viewed and interpreted by the chromatographer using the original processing method that may require manual placement of the baselines, identification of peaks, reintegration or resetting of integration parameters
- The use of any post-run calculations to further calculate results or mean individual injections to a reportable value.
- Generating a report containing the reportable value of each sample along with other quality information in the report as required by the laboratory's customer. This is shown on the right hand side of Figure 2 as the interpretation, calculation and reporting phase of the diagram.
- In addition, audit trail entries are required throughout the whole chromatographic process to identify who did what and when and assure the integrity of the raw data and final results.
You will remember that I asked you to put a thought about the phrase in EU GMP Chapter 4 about raw data that is used to create other records on the back burner. Now we can bring this phrase into the debate and see what this means in terms of a CDS. The AGIT guidance document is similar to the definition of electronic records for a CDS that I outlined in my Questions of Quality column in 2000 (3). Therefore, I have taken the principles of the AGIT Guidance and drawn my interpretation of the electronic raw data for a CDS, which is shown in Figure 2.

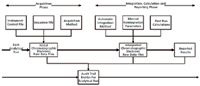
Figure 2: Electronic raw data files for a CDS.
The left hand side of Figure 2 (acquisition phase) can be considered the raw data and the interpretation on the right hand side (integration, calculation and reporting phase) are the "other records". However, a better example is where the CDS is used to create the raw data and the output from the system is then entered into a spreadsheet to calculate further information or other records for Chapter 4 (4). Of course, all the files created during the analytical run in the CDS data files, including all applicable audit trail entries, will be electronic records under 21 CFR 11 (2). So we can see that there are similarities between the Chapter 4 and Part 11 in their respective definitions of raw data and electronic records for a CDS.
There are two important areas to consider:
Take Care 1!: Remember that the Swiss guidance (10) is written for GLP laboratories. There is the statement in the section above that intermediate results obtained are not subject to audit trail and does not necessarily have to be stored or maintained. What does the guidance mean here? Imagine that a chromatographer is interpreting the data files from a sequence of injections, especially near the limits of detection or quantification that is performed in a single session. The chromatographer fits baselines and reintegrates the files from the run, possibly several times during a session, to exercise their professional judgement to obtain the best integration. At the end of the session the interpretation with all the audit trail entries relating to the final saved integration parameters is saved and then, in my view, the statement is acceptable. However, in my opinion and contrary to the advice given in this guidance, all electronic data should be kept; see the discussion in more detail in Take Care 2 below.
The integration of a chromatogram is discussed as follows:
The default integration parameters (slope, minimum peak area) of a HPLC method results in an inappropriate integration of the run due to a large number of noise peaks (see first evaluation). The integration parameters were optimized until an acceptable evaluation was obtained resulting in the integration of the relevant peaks only (see second evaluation). After spectroscopic elucidation and co-chromatography with reference items an assignment of the corresponding metabolite fractions was possible (third evaluation). All intermediate results obtained during the first and second evaluations may be discarded, provided they have not been approved or used in follow-up processes.
Take Care 2!: In the quotation from the AGIT guide (10) above, it states that the first and second evaluation results may be discarded. In a GMP environment this is wrong and will result in a serious non-compliance. The reason is that in US GMP §211.194(a) it states that laboratory records include "complete data secured in the course of testing" (15). Therefore if the initial evaluations are deleted this will result in a non-compliance and questioning of the integrity of all data produced by a laboratory, as we shall see in the next section and later when we discuss the US Compliance Programme Guide.
Exhibit 6: An Inspector Calls
If you have wondered if a Star Trek fan works at the FDA, cease wondering because it is true; Compliance Programme Guide 7346.832 (CPG) is the proof (11). This compliance guide was published in 2010 and will be fully effective by May 2012. It covers the new FDA approach to pre approval inspections (PAIs). There are three objectives outlined in the guide and the main one from the perspective of the raw data debate is objective 3, the data integrity audit:
Audit the raw data, hardcopy or electronic, to authenticate the data submitted in the CMC section of the application. Verify that all relevant data were submitted in the CMC section such that CDER product reviewers can rely on the submitted data as complete and accurate.
The inspector will compare raw data, either paper or electronic files, laboratory analyst notebooks and additional information from the laboratory with summary data filed in the CMC (Chemistry, Manufacturing and Controls) section. The CPG states explicitly:
Raw data files should support a conclusion that the data/information in the application is complete and enables an objective analysis by reflecting the full range of data/information about the component or finished product known to the establishment. Examples of a lack of contextual integrity include absences in a submitted chromatographic sequence, suggesting that the application does not fully or accurately represent the components, process, and finished product (11)
Raw data files in electronic systems will be inspected to check that the data are complete. Note also the comment about absences in a submitted chromatographic sequence and you will understand my severe reservations about not retaining the preliminary evaluations outlined in the Swiss AGIT electronic raw data guidance (10). This returns to the comment made by Furman et al. (13) earlier: save everything.
Exhibit 7: GAMP Laboratory Good Practice Guide
The new version of the GAMP Good Practice Guide (GPG) for validation of laboratory computerized systems is entitled Risk-Based Approach to GxP Compliant Laboratory Computerized Systems (12) and is due to be published later this year. Please note that the title has changed in the revison of the document. The GPG has an appendix that describes how to define electronic records to comply with the requirements of 21 CFR 11 and the Scope and Application guidance (2, 7) and also to define the raw data for computerized systems under EU GMP Chapter 4 (5). In doing so the GPG is up to date with the regulations and their interpretation to help chromatographers and analytical scientists working in regulated laboratories.
In addition there is another appendix in the GPG addressing the integrity of laboratory data including the audit trail; the rationale for this is that without an audit trail there is no way to assure the quality of laboratory data when working in either hybrid or electronic modes. Hence the regulatory emphasis on this feature in the new Annex 11 (4, 6) and implicit in the FDA CPG (11) discussed under Exhibit 6 in this column.
The process presented in the GAMP appendix can be outlined as follows:
1. Define the Intended Use of the Chromatography Data System: to determine the nature of the work and the business process supported by the system.
2. Define the System Architecture: to understand if the records are held centrally or on standalone PCs and also what are the instruments controlled by the system? Are there spectra libraries involved or only conventional chromatographic detectors?
3. Understand the Business Processes Automated by the CDS: is this regulated and if so what is the impact of the system on data integrity, product quality and patient safety? Is the system operated in a hybrid mode or fully electronically with electronic signatures?
4. Define the Raw Data/Electronic Records Generated by the System; it is important to realize that the same system, even within the same organization, can generate different raw data and electronic records depending on how it is used and the regulations applicable to its use. It is imperative that any definition of raw data/ electronic records includes the audit trail or audit trails within the specific CDS installed in your laboratory (12).
Summarizing the Exhibits
We have looked at a number of regulations, regulatory guidance documents and industry guidance documents in a debate to define what raw data really means for a chromatography data system. Therefore I would like to summarize the main elements from all the exhibits we have reviewed in this column before coming to a conclusion.
- Exhibit 1: The FDA's Part 11 Scope and Application guidance (7) says that 21 CFR 11 applies to computerized systems that generate records required by the applicable GMP predicate rule. As a CDS requires interpretation of the electronic records generated as part of the analytical process, a system can only be considered a hybrid or fully electronic system. The typewriter interpretation cannot be used.
- Exhibit 2: The detailed rationale of why paper records cannot be raw data for a CDS is explicitly discussed in the FDA's Q&A guidance (8): paper records are not exact and complete or true copies or even complete data secured in the course of testing under US GMP (15).
- Exhibit 3: EU GMP Chapter 4 (5) acknowledges that records generated from the execution of instructions can exist in both hybrid (paper and electronic records) and homogeneous forms (fully electronic). The new version of this regulation requires that raw data to be used in making quality decisions be documented but does not define what are raw data.
- Exhibit 4: To understand what the term raw data encompasses, the definition of original observations from the US GLP regulations (9) was interpreted to include all data necessary for reconstruction of the study report (GLP) or certificate of analysis (GMP). When considering automated instruments all the files generated in the course of an analytical run are essential for the reconstruction of a study and therefore must be raw data.
- Exhibit 5: In the AGIT guidance (10) the debate is extended in more detail for electronic raw data, specifically for chromatography data systems, and came to similar conclusions of all the files necessary to reconstruct the study. I have one major disagreement with the guidance as I think that all files, including the audit trail, are essential to include within the definition of CDS raw data.
- Exhibit 6: My view is reinforced by the FDA CPG 7346.832 (11) as the data integrity audit to be conducted during a pre approval inspection (PAI) is looking for complete data secured in the course of testing (15). Missing data is the start of a serious investigation into suspected falsification and fraud.
- Exhibit 7: To help chromatographers define electronic records/raw data for their CDS systems the second edition of the GAMP Good Practice Guide: Risk-Based Approach to GxP Compliant Laboratory Computerized Systems (12) has an appendix on this subject (Please note that the title has changed in the revison of the document).
Therefore, is there any other conclusion that raw data for a CDS are the electronic files generated during the course of an analytical run? No. The only question left to be asked now: is the CDS being used as a hybrid or homogeneous system?
Dead as a Dodo: My Raw Data are Paper
During the course of presenting the new requirements for the definition of raw data in the new Chapter 4, I have been amazed by the number of laboratories that still continue to define raw data from their CDS systems as paper. Some organizations even go as far as deleting the electronic files from the system; why not save the inspector some time and start drafting the 483 observations now? In my review of the new requirements in Annex 11 and Chapter 4 regulations last year (6), I used this section's heading to indicate that the argument for defining paper as raw data was as dead as a dodo. Paper as raw data, as I have discussed in this column now and previously (1, 3), is an argument based on the initial interpretations of the GLP and GMP regulations from the 1970s. This thinking is perpetuated by quality assurance attitudes in many companies that are too conservative and have not moved with the times and technology. This is 20th century thinking that must be discarded.
Now both the European and American regulatory authorities have equivalent regulations that recognize the need to identify and maintain electronic records from computerized systems. Therefore, if a CDS is used as a hybrid system, the impact is to ensure that both the signed paper printout and the underlying electronic records that generated it are defined as raw data / electronic records and they are maintained and protected throughout the record retention period. However, this is a stupid approach as you have moved from the frying pan to the fire. You have to maintain records in two different and unconnected media and synchronize them. If we go back to the scenario outlined by Furman et al. (13) earlier in this column, where data files need to be reintegrated after some time has elapsed and newly identified peaks integrated, there will be a second signed printout from the CDS. How will two printouts and the raw data files be synchronized? If you read the first set of results how will you know that another, later, set of results also exists that supersedes the paper you have in your hands? You see the potential mess that this could create?
Now is time to bring the raw data debate into the 21st century and finish it once and for all. It is far better, in my view, to work electronically and, only if absolutely necessary, print out the electronically signed report of the results. All electronic records/raw data are now in a single location on a single storage medium on the CDS server. If they are reintegrated and updated the information is in the same location, the reintegrated results and different versions of reports are visible and the current version is easily identified.
An electronic CDS is not without potential issues, but with diligent design and effective IT support it is achievable as many companies have demonstrated. The following needs to be implemented in any CDS system intended for electronic working:
- Fault tolerant server e.g. duplicate power suppliers and processors
- Redundant disk storage with duplicate disk controllers e.g. redundant array of inexpensive disks (RAID) or Storage Area Network (SAN) for secure data storage
- Effective IT backup including considering moving from tape backup to disk for speed and avoidance of tape reading errors and changes in tape format over time
The rationale is to ensure that data are not lost and the system remains operational. Unlike the stars of some FDA warning letters, such as Cambrex Profarmaco, who lost all their chromatographic data when upgrading the CDS application software (18), or Ohm Laboratories, who had not had the time to backup up the CDS records for several months prior to an inspection (19). This will not happen in your laboratories, will it?
Summary
Raw data for CDSs operating a regulated laboratory must be defined as the electronic records such as chromatographic raw data files, instrument control file, processing method, sequence file etc, that constitute the analytical run, including the applicable audit trail entries. Although it may be acceptable to some dinosaurs working in Quality Assurance, to define paper as raw data defies current regulatory regulations, expectation and logic. It only remains to determine if the CDS is operated as a hybrid (electronic records with signed paper reports) or a homogeneous (electronic records with electronically signed reports) system.
Acknowledgement
The author wishes to thank Lorrie Scheussler for advice during preparation of this article.
"Questions of Quality" editor Bob McDowall is Principal at McDowall Consulting, Bromley, Kent, UK. He is also a member of LCGC Europe's Editorial Advisory Board. Direct correspondence about this column should be addressed to "Questions of Quality", LCGC Europe, 4A Bridgegate Pavillion, Chester Business Park, Wrexham Road, Chester CH4 9QH, UK or e-mail Alasdair Matheson, the editor, at amatheson@advanstar.com
References
(1) R.D.McDowall, LC–GC International QOQ, 9 (12), (1996), 790–793.
(2) 21 CFR 11, Electronic Records; Electronic Signatures final rule, 1997.
(3) R.D.McDowall, LC–GC International QOQ, 13 (9), (2000), 648–657.
(4) EU GMP Annex 11 Computerized Systems, 2011.
(5) EU GMP Chapter 4 Documentation, 2011.
(6) R.D.McDowall, LC–GC Europe QOQ, 24 (4), (2011), 208–216.
(7) FDA Guidance for Industry, Part 11 Scope and Application, 2003.
(8) http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm
(9) 21 CFR 58, Good Laboratory Practice for Non-Clinical Laboratory Studies, 1978.
(10) AGIT, Guidelines For The Acquisition And Processing Of Electronic Raw Data In A GLP Environment, 2005.
(11) FDA Compliance Programme Guide 7346.832, Pre Approval Inspections, 2010.
(12) GAMP Good Practice Guide: Risk-Based Approach to GxP Compliant Laboratory Computerized Systems, International Society of Pharmaceutical Engineering (ISPE), Tampa FL, 2012.
(13) W. Furman, T.Layloff and R.Tetzlaff, JAOAC International, 77 (5), (1994),
1314–1318.
(14) Gamble, Weller and L.Withers, Definition of Raw Data, British Association of Research Quality Assurance, November 1994.
(15) 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceutical Products.
(16) GAMP Good Practice Guide, Part 11 Compliant Electronic Records and Signatures, International Society of Pharmaceutical Engineering (ISPE), Tampa FL, 2006.
(17) Principles on Good Laboratory Practice, OECD Series On Principles Of Good Laboratory Practice And Compliance Monitoring Number 1, Organisation of Economic Co-operation and Development (OECD), Paris,1997.
(18) Cambrex Profarmaco warning letter, October 2009.
(19) Ohm Laboratories warning letter, December 2009.








What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






